Download

1 / 15
290 likes | 828 Views
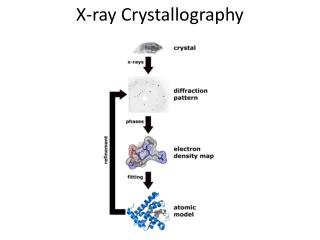
X-Ray Crystallography. Diffraction Phase Problem Crystallization Literature structures. Bragg Equation. n l = 2 d sin q. What we see.

E N D
X-Ray Crystallography • Diffraction • Phase Problem • Crystallization • Literature structures
Bragg Equation nl = 2d sin q
What we see • We measure the intensities and the position of reflections. From the position of the reflection we can determine its index triple (h,k,l) and assign the appropriate intensity to it. There are usually a few ten-thousand reflections collected for each crystal. White spots are reflections
Structure factor F(hkl) This is a sum over all atoms j, with x,y, and z their fractional coordinates. The f (j) is the scattering factor of atom j and depends on the kind of atom and the diffraction angle of the corresponding reflection (h,k,l). For h,k,l =0, f equals the atom's number of electrons. Formula (1) shows that if we know the structure, we can easily calculate structure factors. (1)
Fourier transform • This transformation is accurate and in principle complete. If we know the structure factors we can calculate the actual real structure (the density of the electrons in real space) (2)
The Phase Problem • Unfortunately, a closer look at formula (2) reveals a problem: In order to perform the FT, we need the complex structure factors F(hkl) but we know only their magnitude |Fhkl|. In terms of physics this means that we know only the absolute value of the complex vector F(hkl) but not its phase, a(hkl). This problem is called the Phase Problem. We can reformulate equation (2) as shown below to explicit show the phase, a(hkl). (3)
Solutions to Phase Problem for Proteins (I) • Heavy Atom Method • Substitute with a very heavy atom, which dominates the scattering (and the electron density). Then phase all atoms relative to heavy atom • Drawback is it is difficult to then locate the lighter atoms with any rigor • Substitution can be covalent or non-covalent (solvent)
Solutions to Phase Problem for Proteins (II) • Isomorphous Replacement • Two or more crystals with same structure – can then subtract these differences • Soak protein crystal in solution of different ions, hoping to absorb this ions into the crystal, without changing its shape
Evaluating Correctness of Structures • R-factor • Usually get R<0.06 – smaller the better • Also can look at thermal factors on atomic positions F0 are observed Fc are computed from structure
Crystalization • Crystals need be reasonably large (>0.1 mm in its smallest dimension) so that there is sufficient long range order that an x-ray beam will be diffracted into a discernable pattern of reflections • Start with a solution of the protein with a fairly high concentration (1.5-200 mg/ml) and add reagents that reduce the solubility. By slow concentration, and under conditions suitable for the formation of a few nucleation sites, small crystals may start to grow. Often very many conditions may have to be tried in order to find the proper conditions.
Vapor Diffusion A drop containing protein, stabilizing buffers, and crystallization aids is allowed to equilibrate in a closed system with a much larger reservoir. The reservoir usually contains the same chemicals minus the protein but at an overall higher concentration so that water preferentially evaporates from the drop. If conditions are right this will produce a gradual increase in protein concentration so that a few crystals may form.
Need for Pure Protein • The potential to grow exceptionally good, well ordered protein crystals is there only if very pure protein is available. Proteins less than 95% pure are not likely to produce very good crystals, if they crystallize at all. • Contaminants are: unfolded protein, peptides, particulate matter, anything that causes destabilization of the protein, any chemical that is unnecessary for protein stability/solubility or is present in an unnecessarily high concentration. • The protein solution should contain only those chemicals necessary to maintain protein stability and at the lowest concentrations possible.
Protein Data Bank • http://www.rcsb.org/pdb/ • 41385 Structures • 3-D structures available in interactive form using freeware Chime (www.mdl.com)