Download

1 / 46
740 likes | 1.56k Views
LA COAGULATION. Dr Pierre Hance Marseille HIA Laveran.

E N D
LA COAGULATION Dr Pierre Hance Marseille HIA Laveran
L'hémostase est regroupe un ensemble de processus complexes, et interdépendants, nécessitant la coopération de la paroi vasculaire, des cellules sanguines et des protéines plasmatiques, ayant pour objectif de rétablir le flux sanguin en cas de thrombose , mais également de colmater les fuites pouvant apparaître dans le circuit vasculaire.
Elle se divise en plusieurs étapes: -L’hémostase primaire qui dure de 3 à 5 minutes ,avec formation d’un agrégat plaquettaire. -L’hémostase secondaire , de 5 à 10 minutes, qui permet la consolidation de cet agrégat par de la fibrine. -La fibrinolyse permet en 48 à 72 heures la dégradation du caillot et le retour à une circulation sanguine normale. L'ensemble de ces phénomènes doivent être très finement ajustés pour permettre l'équilibre entre l'hypo et l'hypercoagulabilité.
HEMOSTASE PRIMAIRE I. DEFINITION C’est le temps aboutissant à la formation d’un agrégat plaquettaire ( ou thrombus blanc, ou encore clou plaquettaire.) Elle est suffisante pour arrêter l’hémorragie au niveau des petits vaisseaux, insuffisante mais indispensable pour freiner l’hémorragie au niveau des plus gros vaisseaux. Le clou plaquettaire est fragile et devra être consolidé par le réseau de fibrine au cours de l’hémostase secondaire.
II. Processus: la paroi vasculaire est composée de 3 couches : l'adventice à l'extérieur, la média, et et l'intima, la couche la plus interne. Vont jouer un rôle à ce niveau : - L'endothélium - le facteur Willebrand (Dans le sang, il s'associe au facteur VIIIc (le facteur anti hémophile A) par des ponts Calcium et intervient d'une façon primordiale dans l'adhésivité des plaquettes au sous endothélium) - les plaquettes
III. LE DEROULEMENT DE L’ HEMOSTASE PRIMAIRE A- temps vasculaire: Il y a tout d'abord, au niveau du vaisseau lésé, une vasoconstriction immédiate, tout d'abord passive, lié à l'élasticité de la paroi, puis active, par contraction des fibres musculaires lisses. Cette vasoconstriction peut réduire jusqu'à 30% le calibre du vaisseau lésé, diminuant la fuite sanguine et ralentissant le débit sanguin, ce qui favorise les interactions entre plaquettes et endothélium.
B- temps plaquettaire : B-1- l'adhésion: - Elle s’effectue au sous endothélium en faisant intervenir la membrane plaquettaire B-2- l'activation: Initialement discoïdes, les plaquettes vont devenir sphériques Il se forme alors des ponts de fibrinogène entre les plaquettes, favorisant leur inter adhésion. L’agrégat de plaquettes se forme par apparition successive de couches de plaquettes, réunies les unes aux autres par des ponts fibrinogène; Il se constitue ainsi une sorte de filet qui retient les globules rouges.
IV. EXPLORATION DE L’ HEMOSTASE PRIMAIRE • sur le temps de saignement (Méthode d ’Ivy) (N 1-4 mn) • Devant un allongement du TS, il faut envisager une anomalie d’un ou de plusieurs éléments intervenant dans la physiologie de l’hémostase primaire : • Un trouble plaquettaire quantitatif (thrombopénie) ou qualitatif.( thrombopathie ex : anti agg plaquettaire)) • Une maladie de Willebrand. • Une anémie sévère (Hémoglobine < 80g/l ) • sur la numération plaquettaire ( N 150 à 400 109/l) mais attention aux fausses thrombopénies
V. EXEMPLE DE PATHOLOGIE DE L’HEMOSTASE PRIMAIRE : LA MALADIE DE WILLEBRAND • La maladie de Willebrand est une affection constitutionnelle liée à une anomalie quantitative ou qualitative du facteur Willebrand clinique : hémorragies cutanéo-muqueuses (épistaxis, ecchymoses faciles, ménorragies), hémorragies post-traumatiques, post-opératoires, • Traitement : - En fonction de l ’importance de l ’anomalie, il repose sur l ’utilisation du DDAVP (Minirin), de la corticothérapie.
HEMOSTASE SECONDAIRE Elle a pour expression le passage du sang de l ’état liquide à l ’état de gel par transformation du fibrinogène en une protéine insoluble : la fibrine, qui va former l ’armature du caillot. L ’enzyme responsable de cette transformation est la thrombine
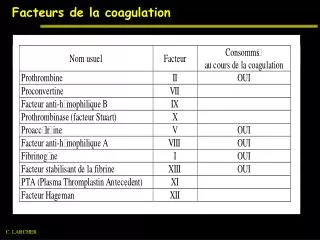
Les facteurs impliqués dans la coagulation : - protéines plasmatiques de la coagulation : environ 10 facteurs(XII, XI, X, IX ou antihémophilique B, VIII ou antihémophilique A, VII, V, II, I. - le facteur tissulaire
Facteurs I fibrinogène (I' = fibrine soluble)(I'' = fibrine insoluble) 120 II prothrombine(IIa = thrombine) 60 III thromboplastine tissulaire ou facteur tissulaire IV calcium V proaccélérine 24 VII proconvertine 6 VIII facteur antihémophilique A 12 IX facteur Christmas ou antihémophilique B 24 X facteur Stuart 40 XI PTA = Plasma Thromboplastin Antecedent 60 XII facteur Hageman 60 XIII facteur stabilisant de la fibrine 150 facteur von Willebrand 24 T1/2(heures)
- les phospholipides - le calcium - les inhibiteurs physiologiques : - Antithrombine III - le système protéine C/ protéine S
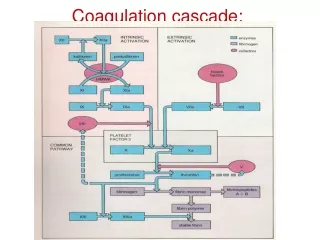
COAGULATION Les différentes étapes : - la formation de la thrombine (selon 2 voies :extrinsèque et intrinsèque). Ces voies font intervenir les différents facteurs de la coagulation qui vont s ’activer en cascade. - la formation de la fibrine - la fibrinolyse (qui assure la disparition du caillot de fibrine)
SCHEMA DE LA COAGULATION F.XII F T-F.VIIa F.XI F.XIa F. IX F. IXa AT PL F. IXa F. VIII F. VIIIa F. VIIIa Ca++ F. VIIa PCa Ca++ F. Xa F. X AT PS PL PL F.Xa F.V F. Va F. Va Ca++ F.II THROMBINE AT Activation THROMBINE-THROMBOMODULINE plaquettaire RAPPELS Le TCA explore : XII, KHPM, PK, XI, IX, VIII, X, V, II, fibrinogène Le TCK n’est pas sensible à l’héparine FIBRINE Le TP explore : VII, X, V, II, fibrinogène Dr Pierre Hance Facteurs Vit K dépendants : II, VII, IX X. Temps thrombine Normal = Pas d’héparine
TCA (intrinsèque/endogène) TQ (extrinsèque) Facteurs contacts FXII FXIIa Facteurs tissulaires +PL+Ca++ FXI FXIa FVIIa FVII FIX FIXa FVIIIa PL Ca++ FXa FVa PL Ca++ FX Voie finale commune Prothrombine Thrombine Fibrinogène Fibrine Fibrinoformation
FIBRINOLYSE La fibrinolyse consiste en la dissolution des caillots intravasculaires par la plasmine. Par ce mécanisme, elle débarrasse la circulation des déchets de fibrine et facilite la reperméabilisation des vaisseaux obstrués par des caillots de fibrine.
EXPLORATION de la COAGULATION • Exploration : - elle se fait après prélèvement sur tube avec anticoagulant (en général bouchon bleu = tube citraté) transporté rapidement au laboratoire ATTENTION au prélèvement : un prélèvement traumatique, un garrot vont modifier les résultats des tests. Noter l ’heure de prélèvement.
Bouchon mauve : EDTA (NF) Bouchon jaune : sec (sérologie) Bouchon bleu : citrate de sodium (coag)
Exploration de la coagulationconditions strictes de prélèvement sanguin • Garrot pose peu de temps • Ponction veineuse franche • Ne pas prélever en premier
4) Rapport anticoagulant /sang correct 5) Hématocrite normal (30 à 50%) 6) Transport entre 10 et 25°C 7) Délai entre prélèvement et analyse < 2H
LES PRINCIPAUX TESTS - Plaquettes sanguines (tube violet EDTA même que NF) - TCK (Temps de céphaline kaolin) : il explore les facteurs XII, XI, IX, VIII, X, V, II et I (N = 31s) - TCA (Temps de céphaline + activateur) : il explore les mêmes facteurs mais est plus sensible à l ’héparine (N = 31s)
LES PRINCIPAUX TESTS • Temps de Quick ou TP (Taux de prothrombine quand exprimé en% : N = 80-100%) et INR. Le TP explore les facteurs, VII ,X, V, II et I. • INR =( TQ malade/TQ témoin)ISI • fibrinogène (N 2-4g/l chez l ’adulte) • dosage des différents facteurs
LES PRINCIPAUX TESTS • Dosage des D-dimères (produits de dégradation spécifiques de la fibrine) • taux élevé dans tous les états d ’activation de la coagulation : CIVD, cancer, infections… • VPN : Diagnostic d ’exclusion d ’une TVP • Dosage des PDF • fibrinolyse
HEMOPHILIE A / B - Affection constitutionnelle, diminution de l ’activité du facteur VIII (Hémophilie A) ou du facteur IX (hémophilie B, mais 6 fois moins fréquente que la A) - Clinique : variable selon l ’importance du déficit, pas d ’accident quand hémophilie modérée - accidents de l ’appareil locomoteur : spontanés, hémarthroses récidivantes, hématomes musculaires - hémorragies gastro-intestinales - hémorragies post-opératoires
HEMOPHILIE A / B - Evolution - séquelles ostéoarticulaires - Traitement - traitement accident hémorragique - traitement substitutif : facteur VIII ou IX - carte d ’hémophile
THROMBOSE Exagération du processus normal de l ’hémostase, qui est déclenché dans la lumière même du vaisseau: - thrombose veineuse : thrombose des membres, thrombophlébites cérébrales, embolie pulmonaire - thrombose artérielle : accident vasculaires cérébraux, infarctus du myocarde
EXPLORATION D ’UNE THROMBOSE • Étiologies principales liées aux anomalies de la coagulation -déficit en AT III - déficit en protéine C - déficit en protéine S - anomalie du facteur V (RPCa) - présence d ’un ACC (anticoagulant circulant) - Il en existe d’autres….(augmentation du F VIII…)
SCHEMA DE LA COAGULATION F.XII F T-F.VIIa F.XI F.XIa F. IX F. IXa AT PL F. IXa F. VIII F. VIIIa F. VIIIa Ca++ F. VIIa PCa Ca++ F. Xa F. X AT PS PL PL F.Xa F.V F. Va F. Va Ca++ F.II THROMBINE AT Activation THROMBINE-THROMBOMODULINE plaquettaire RAPPELS Le TCA explore : XII, KHPM, PK, XI, IX, VIII, X, V, II, fibrinogène Le TCK n’est pas sensible à l’héparine FIBRINE Le TP explore : VII, X, V, II, fibrinogène Dr Pierre Hance Facteurs Vit K dépendants : II, VII, IX X. Temps thrombine Normal = Pas d’héparine
TRAITEMENTS ANTICOAGULANTS Les antagonistes de la vitamine K (AVK): - ils ont pour effet de diminuer la quantité de thrombine formée et sa vitesse de formation - la vitamine K est nécessaire à la synthèse de nombreux facteurs :II, VII, IX, X donc l ’administration d ’AVK va diminuer le taux de ces facteurs et donc diminuer le TP (et donc allonger l ’INR) qui vont servir à surveiller l ’efficacité du traitement: - principaux AVK: Sintron, Previscan….. - leur surdosage entraîne des hémorragies
TRAITEMENTS ANTICOAGULANTS Héparine : médicament de choix pour prévenir et traiter la maladie thromboembolique veineuse On l ’utilise sous deux formes : - non fractionnée (héparine standard) - fractionnée (héparine de bas poids moléculaire)
TRAITEMENTS ANTICOAGULANTS • - Héparine non fractionnée : prévention des risques modérés: 5000 UI 2 ou 3 fois par jour prévention des risques élevés : à adapter quotidiennement pour obtenir un TCA 1,2 à 1,3 fois le témoin (prélever entre 2 inj) traitement curatif d ’une thrombose veineuse constituée : perfusion à la seringue électrique , parfois sous-cutanée, surveillance par TCA entre 2 et 3 fois le témoin, surveillance possible par activité antiXa
TRAITEMENTS ANTICOAGULANTS - Héparine de bas poids moléculaire Fraxiparine, Lovenox, Fragmine, Innohep En traitement préventif pas de surveillance des bilans de coagulation (en dehors du taux de plaquettes a cause de la TIH) En traitement curatif par les HBPM, surveillance par activité anti Xa (prélèvement 3ème ou 4éme heure après l ’injection) Antidote de l ’héparine sulfate de protamine
TRAITEMENTS ANTICOAGULANTS • Le relais d ’un traitement par héparine ou HBPM par les AVK nécessite la prise concomitante des 2 thérapeutiques pendant 5 à 7 jours. L ’héparine ne sera arrêtée qu ’après l ’obtention de deux INR entre 2 et 3.