Download

1 / 28
360 likes | 1.06k Views
Myoglobin and Hemoglobin. Lecture 8, Medical Biochemistry. Lecture 8 Outline. Cooperativity of oxygen binding to hemoglobin Stuctural basis for sickle-cell anemia Modulators of oxygen binding to hemoglobin and the Bohr Effect. Oxygen Saturation Curves for Myoglobin & Hemoglobin.

E N D
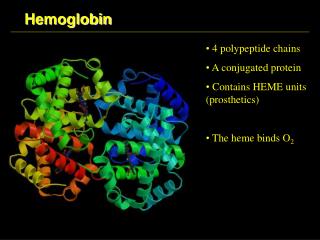
Myoglobin and Hemoglobin Lecture 8, Medical Biochemistry
Lecture 8 Outline • Cooperativity of oxygen binding to hemoglobin • Stuctural basis for sickle-cell anemia • Modulators of oxygen binding to hemoglobin and the Bohr Effect
Oxygen Saturation Curves for Myoglobin & Hemoglobin
Conformational Changes in Hemoglobin upon binding of O2 • The heme iron atom in deoxyHb is slightly out of the plane of the porphryin ring and bound to a histidine imidazole sidechain. This is referred to as a T (“taut”) state by protein chemists • When oxygen binds to the heme on the side opposite the histidine, the iron atom’s electron cloud becomes slightly smaller, and the iron moves into the plane of the porphryin ring, also pulling the histidine side chain in the same direction
Molecular Shift at the Heme Group after Oxygen Binding Blue: deoxy Red: oxy
Conformational Changes (cont) • The movement of the histidine causes the movement of the F-helix. This causes a corresponding rearrangement of the other helices in the protein subunit. The conformational change of the subunit causes it to move away from its partner in the ab pair. Movement of one subunit in the ab pair causes a corresponding conformational adjustment in the paired subunit, enhancing the ability of the latter to bind oxygen.
Dynamics of H-bond and ionic interactions (dotted lines) and van der Waals forces (dashed lines) at the dimer interface when oxygen is bound (blue lines) or unbound (black lines)
Quaternary conformational changes in the oxygen bound (red) and deoxyhemoglobins (blue)
Hemoglobin: Axis of Symmetry Channel View a2 b2 a2 b2 b1 a1 b1 a1 The Oxy form (red) forms a more condensed channel along the interface as compared to the Deoxy form (blue)
Sickle Cell Anemia Sickled RBC Normal RBC Ruptured, sickled RBC
Sickle Cell Anemia • The most common form of sickle cell anemia is caused by a single amino acid substitution of valine for glutamate at position 6 on the b-subunit of hemoglobin. This defect is an autosomal (non-sex chromosome) recessive inherited disease, meaning both parents must be heterozygous carriers to produce a homozygous child. Even heterozygous carriers can experience sickle cell symptoms after vigorous exercise or unpressurized travel at high altitudes.
Sickle Cell Anemia (cont) • The Glu to Val mutation in sickle cell hemoglobin (termed Hb-S) reduces the solubility of deoxyhemoglobin and allows formation of fibrous polymeric filaments of deoxyhemoglobin that precipitate in the red blood cells. This precipitation leads to an ultrastuctural deformity of the red blood cell, the "sickle" shape, which gives these cells a tendency to get hung up and accumulate in the narrow capillaries (thus leading to the associated peripheral pain and many other complications).
Glu to Val Mutation in HbS Position 6 - b-subunits
Subunit interactions that promote polymerization of deoxy-HbS
Sickle Cell Anemia (cont) • Upon exposure of oxygen at the lungs, the HbS filaments immediately dissolve. Thus, situations that slow down flow of blood through the capillaries (normally 0.5 to 2 secs) can lead to the periodic sickle cell anemia “crises”. Conditions like oxygen deprivation (such as high altitudes or strenuous exercise) and dehydration can contribute to slower capillary passage of erythrocytes. The longer times allow deoxy-HbS polymerization to occur, and thus further contribute to the blockage of the affected area.
Other Types of Hemoglobin Mutations (Examples) • Higher O2 affinity, b143 His to Gln; 2,3DPG binding decreased • Lower O2 affinity, b102 Asn to Thr; T-form stabilized • Methemoglobinemias, b63 His to Tyr; increased stability for Fe3+ caused by Tyr residue • a-helix disruptors, b63 His to Pro; Helix E disrupted, heme pocket opened, MetHb forms • Decreased stability of Hb, b43 Phe to Val; heme pocket destabilized
Sickle Cell Anemia - Structure/Function Concepts • Hb-S polymerization illustrates how one amino acid change from a charged to non-polar residue can lead to powerful, cumulative hydrophobic interactions. These are the same forces that hold the Hb monomer cores together, while the salt bridges and hydrogen bonds stabilize the surface interactions between subunits. Loss of the charged surface Glu allows more stable hydrophobic interactions to form in the deoxy-Hb conformation.
2,3 Diphosphoglycerate (DPG) 2,3 Diphosphoglycerate (DPG) (or called 1,3 bisphosphoglycerate (BPG)) binds tightly to the channel interface of deoxyHb, a binding stabilized by interactions with positively charged sidechains. DPG has a poor binding affinity for oxyHb due to the compacted channel
Physiological Function of DPG • The presence of DPG in the erythrocyte creates a dynamic competition for oxygen binding to deoxy-Hb. DPG bound to deoxy-Hb reversibly stabilizes the deoxy conformation. As [DPG] increases, the greater the percentage of oxygen that will be released by Hb at the peripheral tissues. This process occurs naturally due to the differences in oxygen pressures between the lung and periphery, plus the presence of DPG further promotes this oxygen release. This is one mechanism to increase oxygen delivery to tissues during exercise and/or at high altitudes
Model of Hemoglobin Forms “Taut” T-forms, lowest O2 affinity T R “Relaxed” R-forms, highest O2 affinity = DPG
Bohr Effect • Hemoglobin releases H+ when it binds O2 • Hemoglobin binds H+ when it releases O2 • HbO2+ H+HbH+ + O2
Hemoglobin and the Bohr Effect • The Bohr Effect is the direct result of the conformational changes that occur in Hb during oxygen binding. About 50% of the effect is caused by a change in pKa of His 146 (on b subunit). In the T-state, the His is H-bonded to Asp 94. When Hb shifts from the T to R conformation, the H-bond to Asp 94 is disrupted, allowing a proton on the His to readily dissociate. The remainder of the effect results from similar pKa changes of other ionizable groups as a consequence of the T to R transition. This is the O2 bind/H+ release portion of the effect
Hb and the Bohr Effect (cont) • An equivalent amount of H+ released (about 0.31 H+ per oxygen bound) is picked up by Hb as acid (H+) produced from tissue metabolism. • At the lungs, when oxygen rebinds, the H+ released is converted to carbon dioxide and exhaled. About 40% of acid produced by the tissues is buffered in this way by Hb
Hb and Carbon Dioxide Transport • Some of the dissolved CO2 in the blood reacts with amino groups on Hb (and other proteins). This reaction occurs with N-terminal amino groups in the NH2 form; at blood pH, the side chain groups are in the NH3+ form and will not react. About 15% of tissue-produced CO2 is bound by protein in this way (termed carbamino-CO2)
Interactions between DPG, Carbamino-CO2 and Bohr Effect • The carbamino-CO2 can be bound to some of the same groups involved in DPG binding, thus diminishing DPG binding; the reverse can occur, DPG can block CO2 • CO2 also binds to some of the groups involved in the Bohr effect; thus bound CO2 can diminish the H+ buffering capacity of hemoglobin (again the reverse effect can occur, bound H+ blocking CO2