Download

1 / 48
510 likes | 1.24k Views
Myoglobin and hemoglobin. Lecture 3 Dr. Malik ALQUB MD. PHD. Hemoglobin and Myoglobin. First protein to be crystallized - 1849 . First protein to have its mass accurately measured . First protein to be studied by ultracentrifugation. First protein to be associated with function

E N D
Myoglobin and hemoglobin Lecture 3 Dr. Malik ALQUBMD. PHD.
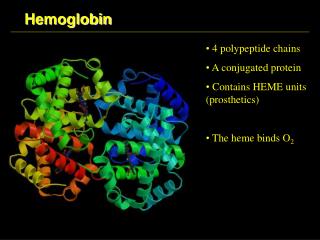
Hemoglobin and Myoglobin • First protein to be crystallized - 1849. • First protein to have its mass accurately measured. • First protein to be studied by ultracentrifugation. • First protein to be associated with function • First protein to show that a point mutation can cause a disease. • First proteins to have his X-ray structure.
The heme group Fe in Mb is Fe2+ - ferrous iron - the form that binds oxygen + Protoporphyrin IX
Overveiw of hemesynthesis Heme Protoporphyrin IX Succinyl CoA + Glycine ALA synthase Protoporphyrinogen IX -aminolevulinic acid Coproporphyrinogen III mitochondrial matrix cytoplasm -aminolevulinic acid Uroporphyrinogen III Porphobilinogen Coproporphyrinogen III Uroporphyrinogen I Coproporphyrinogen I Heme synthesis occurs in all cells due to the requirement for heme as a prosthetic group on enzymes and electron transport chain. By weight, the major locations of heme synthesis are the liver and the erythroid progenitor cells of the bone marrow.
The Conformation Change • Oxygen binding changes the Mb conformation • Without oxygen bound, Fe is out of heme plane • Oxygen binding pulls the Fe into the heme plane • Fe pulls its His F8 ligand along with it • The F helix moves when oxygen binds • Total movement of Fe is 0.029 nm - 0.29 A • This change means little to Mb, but lots to Hb!
How is oxygen carried ? • Complex protein containing heme(iron) • 1 molecule of hemoglobulinecombines with 4 molecules of oxygen to form oxyhaemoglobin Hb + O2 HbO8 Haemoglobin oxygen oxyhaemoglobin This reaction is reversible.
Cooperativity • The haemoglobin molecule is a tetramer composed of 4 protein subunits, each of which can bind a single molecule to O2 • These subunits exhibit cooperativity when they bind to O2 • i.e. the binding of O2 to any subunits increases the likelihood that the other subunits will bind to O2
Oxygen-Hemoglobin Dissociation Curve • Shows the amount of O2 that is bound to haemoglobin (Y-axis) as a function of the partial pressure of O2 in the blood plasma (X-axis). • Because of the cooperativity, this dissociation curve is S-shaped
Plateau: ►haemoglobin highly saturated ► favour the loading of O2 in lung Steep slope: ► small drop of O2 partial pressure leads to a rapid decrease in % saturation of haemoglobin ► favour the release of O2 in tissue cells Body tissue Lung Significance of the S-shape curve 100% % saturation of haemoglobin partial pressure of O2 (mmHg)
Shift to right= low affinity to oxygen 100% % saturation of haemoglobin 50% partial pressure of O2 (mmHg) 26 mmHg
Shift to left= high affinity to oxygen 100% % saturation of haemoglobin 50% partial pressure of O2 (mmHg) 26 mmHg
Binding of CO2 to hemoglobin • Increase the stability of hemoglobin (T form) • Decrease the affinity of hemoglobin to oxygen • Shift the Dissociation Curve to right
Binding of CO to hemoglobin • Increase the (R form) • Increase the affinity of hemoglobin to oxygen
PH and Oxygen-Hemoglobin Dissociation Curve (bohr effect) About 70% of the blood CO2 reacts with H2O to form carbonic acid CO2 + H2O H2CO3 Carbonic acid rapidly dissociates into H2CO3 H+ + HCO-3 ion H+ and bicarbonate ion Carbonic anhydrase
Increased PH and Oxygen-Hemoglobin Dissociation Curve (bohr effect) Ion H+ • Increase the stability of hemoglobin (T form) • Decrease the affinity of hemoglobin to oxygen • Shift the Dissociation Curve to right
Bohr effect – the effect of [PH] on haemoglobin 100% Higher [CO2] e.g. tissue cells ► curve shift to the right % saturation of haemoglobin ► always at a value of lower % saturation of haemoglobin than the curve at the left ► haemoglobin has a lower affinity to O2 partial pressure of O2 (mmHg) ∴ in actively respiring tissue cells, O2 is more easily released by haemoglobin !
Shift to right = low affinity to oxygen 100% Increased CO2 decreased PH Increased 2.3 BPG % saturation of haemoglobin 50% partial pressure of O2 (mmHg) 26 mmHg
Shift to left= high affinity to oxygen 100% decresed CO2 increased PH decresed 2.3 BPG % saturation of haemoglobin 50% partial pressure of O2 (mmHg) 26 mmHg
An Erythrocyte (RBC) Normal Values RBCs, male 4.7-6.1 x 106/µL female 4.2-5.4 x 106/µL Hb, male 13.0-16.0 g/dL female 12.0-15.0 g/dL Hct, male 42-53% female 37-47% MCH 29±2 pg MCV 81-94 fL MCHC 32-37.5% Practical Values 65% of Fe in Hb 1 g Hb = 3.46 mg Fe 1 mL blood at 15 g/dL Hb = 0.5 mg Fe RBC x 3 = Hb Hb x 3 = Hct Microcytic < 81 fL Macrocytic > 94 fL
Hemoglobin A1c • Diabetes • Increased glucose concentration • Nonenzymaticglycosylation • Associated with diabetic complication
Genetics. • In the first 8 weeks of embryonic life the predominant forms of hemoglobin are: • Hb Gower 1 (ζ2ε2). • Hb Gower 2 (α2ε2). • Hb Portland 1 (ζ2γ2). • By the 12th week embryonic hemoglobin is replaced by Hb F (α2γ2) which represents 70 – 100% of hemoglobin in fetal life.
Genetics (2). • Adult hemoglobin Hb A (α2β2) detectable from 16/40, replaces Hb F as predominant hemoglobin by 6/12 after birth, up to 30% of Hb in fetal life. • In normal adults 96 – 98% of hemoglobin is HbA, HbF(<1%) constitute a minor component of the total hemoglobin.
Fetalhemoglobin • Is the mainoxygen transport proteinin thefetusduring the last seven months of development in theuterusand in the newborn until roughly 6 months old. • Baby takes about 2 years to completely switch over to adult haemoglobin
Function • Fetal hemoglobin differs most fromadult hemoglobinin that it is able to bind oxygen with greater affinity than the adult form, giving the developing fetus better access to oxygen from the mother'sblood stream. • The transfer of oxygen is from the mother (less tightly bond) to the baby (more tightly bond).
Characteristic • Hemoglobin F is made up of 2 alpha chains and 2 gamma chains • Hemoglobin F does not turn into hemoglobin A. • Hb F and Hb A are completely different hemoglobin • Newborn babies with sickle cell disease make hemoglobin F and hemoglobin S
Sickle cell anemia • Hemoglobin molecules in each red blood cell carry oxygen from the lungs to body organs and tissues and bring carbon dioxide back to the lungs. • In sickle cell anemia, the hemoglobin is defective. After hemoglobin molecules give up their oxygen, some may cluster together and form long, rod-like structures. These structures cause red blood cells to become stiff and assume a sickle shape.
Characters of Sickled Red Cell • Changing the shape of the RBC from a round disc to a characteristic crescent (sickle) shape. • Sickled red cells cannot squeeze through small blood vessels. • They stack up and cause blockages that deprive organs and tissues of oxygen-carrying blood. • This process produces periodic episodes of pain and ultimately can damage tissues and vital organs and lead to other serious medical problems.
Characters of Sickled Red Cell • Normal red blood cells live about 120 days in the bloodstream, but sickled red cells die after about 10 to 20 days. • Because they cannot be replaced fast enough, the blood is chronically short of red blood cells, a condition called anemia.
Inheritance • Sickle cell anemia is an autosomal recessive genetic disorder caused by a defect in the HBB gene, which codes for hemoglobin. • The presence of two defective genes (SS) is needed for sickle cell anemia. • Hemoglobin S differs from normal adult hemoglobin (hemoglobin A) only by a single amino acid substitution (a valine replacing a glutamine in the 6th position of the beta chain of globing). • When a person has two copies of the S gene (homozygous SS), he has sickle cell anemia.
Inheritance • In sickle cell disease, as much as 80% to 100% of the hemoglobin may be HbS. • A person with one altered S gene will have sickle cell trait. • In those who have sickle cell trait, 20% to 40% of the hemoglobin is HbS. • The person does not generally have any symptoms or health problems but can pass the gene on to his children.
Inheritance • If each parent carries one sickle hemoglobin gene (S) and one normal gene (A), each child has a 25% chance of inheriting two defective genes and having sickle cell anemia. • 25% chance of inheriting two normal genes and not having the disease. • 50% chance of being an unaffected carrier like the parents.
Pathophysiology • Normal hemoglobin exists as solitary units whether oxygenated or deoxygenated (upper panel). • In contrast, sickle hemoglobin molecules adhere when they are deoxygenated, forming sickle hemoglobin polymers (lower panel).
Hemoglobin C • This results when one of the beta subunits is replaced with beta C The mutation that causes this change in the beta happens because a glutamic acid residue replaces a lysine residue at the sixth position of the beta globin chain.
Hemoglobin Electrophoresis Start position - + HbA HbS HbC - + anode cathode migration
Beta Thalassemia • βthal major is homozygosity or compound heterozygosity resulting in severe phenotype. • βthal minor or trait is heterozygosity with asymptomatic phenotype.
Alpha Thalassemia α α α α Father Mother