Download
1 / 60
610 likes | 942 Views
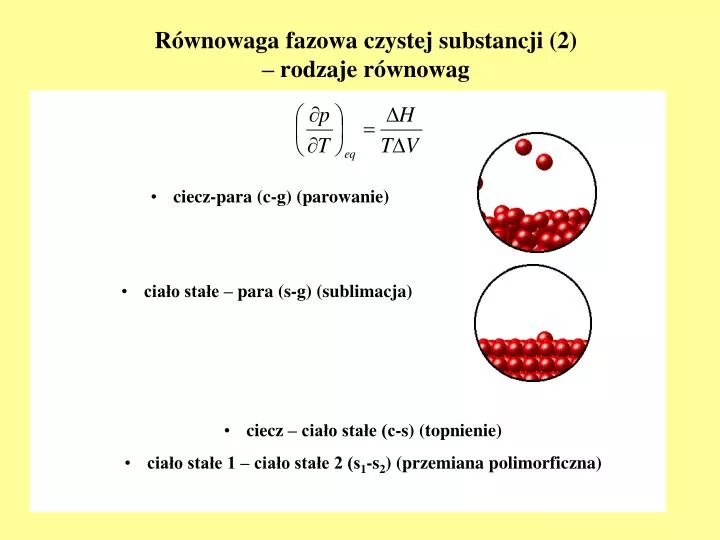
Równowaga fazowa czystej substancji (2) – rodzaje równowag. ciecz-para (c-g) (parowanie). ciało stałe – para (s-g) (sublimacja). ciecz – ciało stałe (c-s) (topnienie) ciało stałe 1 – ciało stałe 2 (s 1 -s 2 ) (przemiana polimorficzna). Zależności jakościowe p ( T ).

E N D
Równowaga fazowa czystej substancji (2)– rodzaje równowag • ciecz-para (c-g) (parowanie) • ciało stałe – para (s-g) (sublimacja) • ciecz – ciało stałe (c-s) (topnienie) • ciało stałe 1 – ciało stałe 2 (s1-s2) (przemiana polimorficzna)
Zależności jakościowe p(T) Równowaga fazowa czystej substancji (9) p(T) – funkcja rosnąca p(T) – funkcja rosnąca p(T) – funkcja rosnąca p(T) – funkcja malejąca
Równowaga fazowa czystej substancji (3) – równowaga parowania (1) Najbardziej radykalne uproszczenie – równanie Clausiusa-Clapeyrona • Entalpia parowania nie jest funkcją temperatury (i w związku z tym i ciśnienia). • Faza gazowa jest gazem doskonałym, czyli jej objętość molowa wyraża się wzorem • Objętość fazy ciekłej jest znacznie mniejsza od objętości fazy gazowej i może być pominięta. Umożliwia to zastąpienie zmiany objętości poprzez objętość pary
Równowaga fazowa czystej substancji (4) – równowaga parowania (2) równanie Clausiusa-Clapeyrona
równanie Clausiusa-Clapeyrona Równowaga fazowa czystej substancji (5)– równowaga parowania (3) Założenia: a. Entalpia parowania nie jest funkcją temperatury (i w związku z tym i ciśnienia). b. Faza gazowa jest gazem doskonałym c. Objętość fazy ciekłej jest znacznie mniejsza od objętości fazy gazowej i może być pominięta. Równanie Clausiusa-Clapeyrona zawodzi dla: • Warunków bliskich krytycznym. 2. Dużych różnic temperatury.
modyfikacja równania Clausiusa-Clapeyrona Równowaga fazowa czystej substancji (6) – równowaga parowania (4) równanie Antoine’a
równanie Clausiusa-Clapeyrona Równowaga fazowa czystej substancji (7) – równowaga sublimacji (3) Równanie Clausiusa-Clapeyrona zupełnie nieźle opisuje równowagę sublimacji
niezależne od T,p Równowaga fazowa czystej substancji (8) – równowaga topnienia (1) Analogiczne równanie otrzymuje się dla równowagi pomiędzy odmianami polimorficznymi
p Typowy diagram fazowy p,T płyn nadkrytyczny k. topnienia A K s c p=1 atm P3 k. parowania g B T k. sublimacji
p płyn nadkrytyczny K Typowy diagram fazowy p,V c s+c c + g s g p3 s + g V Vs Vc Vg
Typowy diagram fazowy p,V,T S. Stølen, T. Grande, N.L. Allan, Chemical Thermodynamics of Materials, J. Wiley & Sons, Ltd, 2004
Diagram fazowy p,T wody W. P. Atkins, Chemia fizyczna, PWN, 2001
Diagram fazowy p,V,T wody pod wysokim ciśnieniem H. Buchowski, W. Ufnalski, Fizykochemia gazów i cieczy, WNT, 1998
Diagram fazowy p,V,T siarki H. Buchowski, W. Ufnalski, Fizykochemia gazów i cieczy, WNT, 1998
Potencjał chemiczny czystego gazu doskonałego w funkcji ciśnienia dla czystej substancji Potencjał chemiczny składników w mieszaninie (1)
Potencjał chemiczny składnika w mieszaninie gazów doskonałych Potencjał chemiczny składników w mieszaninie (2) pi = pxi → ciśnienie cząstkowe ciśnienie cząstkowe dla gazu doskonałego
Potencjał chemiczny składnika w mieszaninie rzeczywistej (1) Potencjał chemiczny składników w mieszaninie (3) fi = lotność p→ 0 fi→ pi ϕi = fi /pi fi = piϕi ϕi -współczynnik lotności p→ 0 ϕi → 1
Potencjał chemiczny składnika w mieszaninie rzeczywistej (2) Potencjał chemiczny składników w mieszaninie (4) Lotność (współczynnik lotności) można obliczyć na podstawie równania stanu
Roztwór doskonały (1) Potencjał chemiczny składnika w mieszaninie gazów doskonałych Potencjał chemiczny składników w mieszaninie (5) Fenomenologiczna definicja roztworu doskonałego: Roztwór doskonały to mieszanina, w której potencjały chemiczne wszystkich składników wyrażają się powyższym wzorem.
Roztwór doskonały (2) Interpretacja molekularna roztworu doskonałego: Mieszanina, w której właściwości różnych cząsteczek (oddziaływania i entropie) są takie same Potencjał chemiczny składników w mieszaninie (6) Mieszaniny bliskie doskonałości: benzen + toluen, H2O+ D2O, 1-propanol + 2-propanol Mieszaniny o dużych odchyleniach od doskonałości: H2O + etanol, n-heksan + kwas octowy
Potencjał chemiczny składników w mieszaninie (7) ai=aktywność ai = 1 dla i(T, p) = io(T, p) γi = ai /xi Aktywności można wyrazić poprzez współczynniki aktywności ai = xiγi Różne sposoby definiowania stanu odniesienia
Sposoby definiowania współczynników aktywności („systemy odniesienia”) Symetryczny system odniesienia Dla każdego składnika: xi→ 1 γi→ 1 i(T,p) = io(T,p)+ RTln(xiγi) io(T,p) – potencjał chemiczny czystego składnika Potencjał chemiczny składników w mieszaninie (8) Niesymetryczny system odniesienia Dla składnika (składników) występujących w rozcieńczeniu xi→ 0 γi*→ 1 i(T,p) = io(T,p)+ RTln(xiγi*) io(T,p) – potencjał chemiczny składnika „w rozcieńczeniu nieskończenie wielkim, ekstrapolowany do czystej substancji” Pozostałe składniki (występujące w nadmiarze) – definicja jak w symetrycznym układzie odniesienia
μi μio duże stężenia Jeszcze coś o współczynnikach aktywności (1) μiid = μio +RTlnxi μi* μi = μio +RTln(xiγi) ideal RTlnxi = μi* +RTln(xiγi*) -∞ 0 μi = μi* +RTlnxi duże rozcieńczenia i* = i /i
lnγA,lnγB lnγB∞ Jeszcze coś o współczynnikach aktywności (2) lnγA∞ lnγB lnγA 0 xB lnγA* i* = i /i lnγB* A B lnγi* =lnγi- lnγi∞
Nie zachodzą, bo np. niemożliwa jest reakcja H2O + N2 → CO2 + Fe Dlaczego pewne reakcje zachodzą, a inne nie? Ale także niemożliwy jest proces N2 + H2 → 2NH3 - są to ograniczenia stechiometryczne (bilansowe) (1) Niemożliwe są procesy, które nie spełniają wymogu minimalizacji (maksymalizacji) odpowiedniego potencjału termodynamicznego – - są to ograniczenia termodynamiczne (2) Praktycznie nie zachodzą procesy bardzo powolne - są to ograniczenia kinetyczne (3)
Proces nie narusza ograniczeń termodynamicznych • – nie gwarantuje to jego zachodzenia. • Proces narusza ograniczenia termodynamiczne • – oznacza to, że na pewno nie zajdzie. Ograniczenia termodynamiczne na pewno nie zajdzie!
potencjały chemiczne dG = -SdT + Vdp + idni Różniczka zupełna G: zmienna uniwersalna, niezależna od i Różniczka zupełna G w układzie otwartym (1) Niezależne parametry: p, T, n1, n2,…,nk? Ale przecież mamy prawo stosunków wielokrotnych… Zmienna reakcji (współrzędna reakcji, liczba postępu reakcji)
Zmienna reakcji (1) Ponieważ liczbę moli każdego reagenta da się przedstawić jako: jedynym niezależnym parametrem określającym ilości składników jest ξ. Mamy zatem G(T,p, ξ), a nie G(T,p,n1,n2,…,nk) !
znika przynajmniej jeden substrat znika przynajmniej jeden produkt Zmienna reakcji (2) 1. Dla początku reakcji (ni= nio), ξ = 0. 2. ξ,reakcja zachodzi w prawo; ξ ,reakcja zachodzi w lewo. 3. ξmoże się zmieniać tylko w ściśle określonych granicach: ξmin≤ ξ ≤ ξmax
Wyznaczanie zakresu zmienności ξ - przykład Zmienna reakcji (3) 2HCl(g) + COCl2(g) = H2O(g) + C(grafit) + 2Cl2(g) początkowe liczby moli reagentów: ξmax 2,5 2 -0,5 ≤ ξ ≤ 2 -3 ξmin brakuje Cl2 -2 brakuje COCl2 -0,5 ξmin 0 -3 -2 -0,5 2 2,5 ξmax
Zmienna reakcji (1) Ponieważ liczbę moli każdego reagenta da się przedstawić jako: jedynym niezależnym parametrem określającym ilości składników jest ξ. Mamy zatem G(T,p, ξ), a nie G(T,p,n1,n2,…,nk) !
dG = -SdT + Vdp + idni Różniczka zupełna G: Różniczka zupełna G w układzie otwartym (2) Niezależne parametry: p, T, ξ dG = -SdT + Vdp + iidξ (ii )dξ entalpia swobodna reakcji dG(T,p,ξ) = -SdT + Vdp + (ii)dξ
dG(T,p,ξ) = -SdT + Vdp + (ii)dξ Warunki zachodzenia reakcji chemicznej (1) T,p = const → dG(T,p,ξ) = (ii)dξ dG < 0 reakcja zachodzi → ii < 0 dG > 0 reakcja zachodzi ← ii > 0 dG = 0 równowaga ii = 0
Wyrażenie na entalpię swobodną reakcji: Warunki zachodzenia reakcji chemicznej (2) i (T,p,xi) = io(T,po) + RTlnai gdzie ai= - dla gazów ale dla gazów doskonałych ai = pxi/po 1 - czysta substancja skondensowana xiγi - składnik w roztworze ciekłym lub stałym ale dla roztworu doskonałego ai = xi
i = io + RTlnai (∂G/∂ξ)T,p = ii = iio + RT ilnai(ξ) Warunki zachodzenia reakcji chemicznej (3) ∆Go ∆Go = iio standardowa entalpia swobodna reakcji reakcja zachodzi ∆Go + RT ilnai(ξ)< 0 reakcja zachodzi ∆Go + RT ilnai(ξ)> 0 równowaga ∆Go + RT ilnai(ξ)= 0
∆Go + RT ilnai = 0 Warunki zachodzenia reakcji chemicznej (4) K - stała równowagi iloraz reakcji Analogicznie „>” dla ← i „<„ dla →
reakcja zachodzi → reakcja zachodzi ← równowaga Warunki zachodzenia reakcji chemicznej (5) Istnienie (albo nie) równowagi oraz kierunek zachodzenia procesu może być interpretowany jako wynik relacji między ilorazem reakcji a stałą równowagi
K = exp(-∆Go/RT) Parę słów o stałej równowagi 1. Stała równowagi nie jest bytem koniecznym (jak ∆Go), jakkolwiek czasami użytecznym. 2. Zależy tylko od temperatury. 3. Nie zależy od stężenia (!!!), a tylko w stanie równowagi równa się ilorazowi reakcji.
Stała równowagi K = exp(-∆Go/RT)
Warunek równowagi chemicznej Iloraz reakcji jest funkcją ξ, bo ai zależy od xi, któryzależy od ξ ai= - dla gazów 1 - czysta substancja skondensowana xiγi - składnik w roztworze ciekłym lub stałym
1. Określenie stanu równowagi możliwe jest przy ustalonych 2 parametrach lub narzuconych więzach na układ (np. p, T = const), ale mogą być też inne: (V, T = const), (Q = 0, p = const), (Q = 0, V = const). Przy mniejszej liczbie więzów, stan równowagi nie zostanie osiągnięty (!). Jak znaleźć położenie stanu równowagi (1) ? 2. Podstawowy parametr, który określa położenie stanu równowagi, to zmienna reakcji ξ. Znaleźć położenie stanu równowagi, oznacza znaleźć wartość ξ(i wynikające stąd ilości reagentów). 3. Ponieważ w stanie równowagi musi być spełnione równanie lub jego alternatywna forma: ∆Go + RT ilnai(ξ)= 0, podstawowy algorytm polega na rozwiązaniu któregoś z tych równań względem ξ
Jeszcze raz to samo Co ? Znaleźć położenie stanu równowagi Jak znaleźć położenie stanu równowagi (2) ? Jak ? Rozwiązać poniższe równanie względem ξ To właśnie równanie należy rozwiązać względem zmiennej reakcji (ξ)
Możliwe algorytmy znajdowania ξ* G (∂G/∂ξ)T,p Ilustracja graficzna Bezpośrednie znajdowanie minimum G (metody optymalizacji !!) Rozwiązanie równania względem ξ ξ = 0 ξ = ξ* ξ = ξ* ξmin ξmin ξ = 0 ξmax ξmax
3. Rozwiązać poniższe równanie względem ξ Algorytm obliczeń 1. Obliczyć stałą równowagi. 2. Wyrazić iloraz reakcji jako funkcjęξ. Zwykle kolejność jest następująca: 2.1. Najpierw znajduje się zależność ilorazu reakcji od ułamków molowych. 2.2. Następnie wyraża się xi poprzez ξ.
Standardowa entalpia swobodna reakcji Standardowa entropia reakcji Jak wyznaczyć stałą równowagi (1) ∆G0 = ∆H0 - T∆S0 (bo: G = H – TS) Standardowa entalpia reakcji Obliczanie ∆H0 Obliczanie ∆S0 Potrzebne dla każdego reagenta
ai = pxi/po 2N2(g) + O2(g) = 2N2O(g) Przykład z życia (1) Dla niskich i umiarkowanych ciśnień: Wyrażamy xijako funkcję ξ zwykle = 0 (produkt) n1o - 2ξ (n1o - 2ξ)/ ∑ni n2o - ξ (n2o – ξ)/ ∑ni n3o + 2ξ (n3o + 2ξ)/ ∑ni ∑ni = n1o + n2o + n3o - ξ
2N2(g) + O2(g) = 2N2O(g) Przykład z życia (2) n1o - 2ξ (n1o - 2ξ)/ ∑ni n2o - ξ (n2o – ξ)/ ∑ni Uwaga na wielość pierwiastków! n3o + 2ξ (n3o + 2ξ)/ ∑ni ∑ni = n1o + n2o + n3o - ξ Rozwiązać to równanie względemξ
reakcja w lewo ← > < = = K Wpływ różnych czynników na położenie stanu równowagi (T, p, gaz obojętny) reakcja w prawo → brak wpływu 1. Układ jest w stanie równowagi. 2. Stan równowagi ulega zaburzeniu pod wpływem jakiegoś czynnika. 3. Co wymusza określony bieg reakcji aż do osiągnięcia ponownego stanu równowagi.
Wpływ temperatury na położenie stanu równowagi (1) 1. Od temperatury zależy silnie prawa strona (K). 2. Wpływ temperatury na iloraz reakcji (poprzez współczynniki lotności lub aktywności) jest znikomy. 3. Do wyjaśnienia – jak K zależy od temperatury?
Zależność stałej równowagi od temperatury (1) zależy od T poprzez Δcp0 ΔG0 zależy od temperatury, bo ΔG0(T) = ΔH0(T) - TΔS0(T) Do wyjaśnienia – jak ΔG0 (lub G) zależy od temperatury?






















