Download

1 / 47
470 likes | 599 Views
Curso de Introdução à Bioinformática. Programa de Qualificação Docente da CAPES Convênio: UFPE - UFCG - Fiocruz. Comparação de Seqüências e Busca por Similaridade. Marcos Catanho. Laboratório de Genômica Funcional e Bioinformática DBBM-IOC / Fiocruz. Agenda. Motivação Métodos utilizados

E N D
Curso de Introdução à Bioinformática Programa de Qualificação Docente da CAPES Convênio: UFPE - UFCG - Fiocruz Comparação de Seqüências e Busca por Similaridade Marcos Catanho Laboratório de Genômica Funcional e Bioinformática DBBM-IOC / Fiocruz
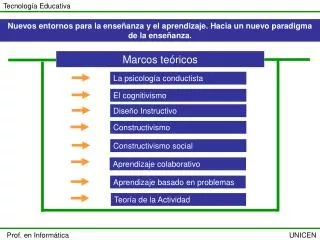
Agenda • Motivação • Métodos utilizados • Problemas freqüentes • Heurística para alinhamento local (BLAST) • Pacotes de programas para análise de seqüências
Os motivos... • Comparação entre seqüências biológicas (ácidos nucléicos e proteínas) uma das tarefas computacionais mais freqüentes entre pesquisadores da área biológica.
Os motivos... • Este tipo de análise permite que relações evolutivas, estruturais e funcionais existentes entre as seqüências comparadas sejam reveladas, fornecendo evidências decisivas para a caracterização das propriedades biológicas de novas seqüências com base no conhecimento acumulado sobre outras já estudadas.
Os motivos... • Se as seqüências comparadas são suficientementesimilares entre si, infere-se que estas seqüências sejam homólogas. • E sendo assim, presume-se que elas tenham a mesma estrutura e função biológica.
Os problemas... • Qual o grau de similaridade mínimo entre duas seqüências para que possam ser consideradas homólogas? • Similaridade ao nível da seqüência primária por si só é suficiente para determinar se duas seqüências são homólogas ou não?
Os problemas... • O que fazer quando a seqüência de função desconhecida é bastante similar a várias seqüências de um outro organismo, mas que exercem funções diferentes no mesmo? Qual delas é o “verdadeiro homólogo”? O mais similar? • Já foi demonstrado que muitas vezes não é... “E agora José?” • Portanto, em princípio, similaridade não implica necessariamente em homologia.
Nem tudo está perdido... Será? • Atualmente, os algoritmos de comparação de seqüências são acompanhados por estimativas estatísticas que fornecem uma medida do grau de significância das similaridades observadas, auxiliando a dedução de homologia. • Mesmo assim, significado estatístico não implica necessariamente em significado biológico...
(Parênteses - algoritmo) • É um conjunto de instruções ordenadas para execução de uma ação qualquer.
Os métodos... • Durante a evolução, as seqüências mudam através de inserções, deleções e mutações. • Estes eventos podem ser traçados com uso de algoritmos de alinhamento.
Os métodos... • Por exemplo, suponha que a seqüência a tenha evoluído para a seqüência b, através de inserções, substituições e deleções. Podemos representar esta transformação da seguinte maneira: a = A C - T T G A b = A G A T T - A
Os métodos... • O objetivo é achar o alinhamento “correto” que representa a verdadeira série de eventos evolutivos que ocorreram.
Os métodos... • Para cada alinhamento, calcula-se o número de pontos obtidos (score), com base em um esquema de pontuação (ou matriz de substituição) e em valores arbitrados de penalidade para a abertura e extensão de espaços nas seqüências alinhadas (gap opening/extension penalties).
Os métodos... a = A C - T T G A b = A G A T T - A pontuação = 1 0 -1 1 1 -1 1 score = 1+0+(-1)+1+1+(-1)+1 = 2 Esquema de pontuação: match = 1 mismatch = 0 gap = -1
(Parênteses – matriz de substituição) • É uma matriz representando todas as possíveis trocas entre aminoácidos, onde um valor é atribuído a cada uma destas trocas. • Esses valores são proporcionais à probabilidade de ocorrência de cada troca, tomando-se como base um determinado modelo evolutivo.
(Parênteses – matrizes de substituição) • PAM (Percent Accepted Mutation) family: • Baseiam-se em alinhamentos globais de proteínas muito próximas • PAM1 é a matriz calculada a partir da comparação de seqüências com não mais do que 1% de divergência • As demais matrizes PAM são extrapolações da PAM1
(Parênteses – matrizes de substituição) • BLOSUM (BLOcks SUbstitution Matrix) family: • Baseiam-se em alinhamentos locais de proteínas • BLOSUM 62 é a matriz calculada a partir da comparação de seqüências com não menos do que 62% de divergência • Todas as matrizes BLOSUM baseiam-se em alinhamentos observados; não há extrapolações
Dica importante! • Seqüências que codifiquem proteínas ou que potencialmente codifiquem proteínas devem ser alinhadas na forma de aminoácidos e não de nucleotídeos. • Motivos: • Maior precisão (por causa da degeneração do código genético) • Maior sensibilidade (leva em conta características físico-químicas dos aminoácidos)
Os métodos... • Alinhamento global • Alinhamento de pares de seqüências nucleotídicas ou protéicas ao longo de toda a extensão das mesmas. • Apropriado nos casos em que se espera que as seqüências estudadas sejam similares ao longo de toda a seqüência ou na maior parte dela.
Os métodos... • Alinhamento local • Alinhamento de uma ou mais partes de duas seqüências nucleotídicas ou protéicas. • Apropriado nos casos em que se espera que apenas algumas regiões específicas das seqüências estudadas (e.g domínios) sejam similares entre si. • Neste caso, o alinhamento global das seqüências poderia não ser apropriado (as similaridades locais poderiam ser “mascaradas”)
Os métodos... • Rigorous Dynamic Programming • Needleman & Wunsch (1970) (global) • Smith & Waterman (1981) (local) SSEARCH • Heuristics • Lipman & Pearson (1985,1988) (local) FASTA • Altschul et al. (1990,1997) (local) BLAST • Feng & Doolittle (1987) (global) • Thompson et al. (1994) (global) ClustalW
BLAST - Basic Local Alignment Search Tool • Provavelmente a ferramenta computacional mais utilizada em biologia molecular e bioinformática • Busca seqüências armazenadas nos bancos de dados pela similaridade entre a estrutura primária da seqüência query e as seqüências armazenadas no banco
BLAST - Basic Local Alignment Search Tool • Propriedades biológicas descritas para seqüências armazenadas podem ser transferidas para a seqüência query desde que suas estruturas primárias sejam semelhantes • O maior problema é definir um cut-off, um limite abaixo do qual as similaridades encontradas entre a query e os hits não sejam mais significativos
BLAST - Basic Local Alignment Search Tool • É um método heurístico para alinhamentos locais • Projetado especialmente para buscas em bancos de dados • Idéia básica: bons alinhamentos irão conter pequenos trechos de combinações iguais
BLAST - algoritmo • 1. Filtrar as regiões de baixa complexidade FTLPQITTPPITTPPLTIDPINLTGFTLPQITTPPITTPPLFN ||| FTLPQ(ITTPP)2LTIDPINLTGFTLPQ(ITTPP)2LFN FTLPQXXXXXXXXXXLTIDPINLTGFTLPQXXXXXXXXXXLFN
BLAST - algoritmo • 2. Fragmentar a seqüência query e as seqüências depositadas no banco de dados, criando “palavras“ (de comprimento 3 para proteínas e 11 para DNA) através do uso de uma janela deslizante MEFPGLGSLGTSEPLPQFVDPALVSS MEF EFP FPG PGL GLG
BLAST - algoritmo • 3. Utilizando uma matriz de substituição (pontuação) (PAM, BLOSUM), encontrar todas as “palavras” de tamanho W que obtenham, no mínimo, um no. de pontos (score) T quando comparadas com a seqüência query, criando uma lista de “palavras” de alta pontuação
BLAST - algoritmo • 4. Procurar em cada seqüência depositada no banco de dados por uma ou mais ocorrências de cada “palavra” de alta pontuação. Cada uma destas ocorrências (hit) será uma “semente” para um alinhamento sem gaps • 5. Estender os hits em ambas as direções, na tentativa de gerar alinhamentos com score acima de um limiar S
BLAST - algoritmo • 5.1. BLAST original (ungapped): extensão dos hits à esquerda e à direita da “semente”, sem gaps. Esta extensão irá continuar enquanto o score aumentar ou pelo menos continuar o mesmo. O alinhamento obtido é chamado HSP (High Scoring Pair) • 5.2. Atualmente (gapped): hits ao longo da mesma diagonal (Dot plot) com uma distância A entre os dois são reunidos e a extensão se dá com a seqüência maior obtida
(Parênteses – Dot Plots - matrizes de homologia) Auto-comparação do receptor de LDL humano. A: janela = 1, estringência = 1 B: janela = 23, estringência = 7
BLAST - algoritmo • 6. Reter somente os HSPs com score acima do limiar S • 7. Determinar a significância estatística de cada alinhamento remanescente (p-value e E-value) • 8. Mostrar os alinhamentos locais (de acordo com Smith-Waterman)
Resultado (input) • Seqüência em formato FASTA • Primeira linha: cabeçalho (header) • Demais linhas: seqüência Símbolo “>” Nome da seqüência Descrição da seqüência
Resultado (BLASTN) • O output é dividido em cinco partes: • 1. Header contendo a versão do BLAST, data da compilação, referência, RID, etc. • 2. Representação gráfica dos alinhamentos • 3. Sumário com uma descrição em uma linha de cada hit • 4. Os alinhamentos com seus respectivos parâmetros calculados • 5. Rodapé com a descrição detalhada dos parâmetros de busca empregados, o banco de dados, etc.
Resultado (links) • G: Gene (banco de dados de genes) • U: UniGene (banco de dados de clusters de genes) • E: GEO Profile (dados de expressão gênica e hibridização genômica obtidos por tecnologia high-throughput)
Pacotes de programas para análise de seqüências • GCG (pago) • http://www.accelrys.com/products/gcg/ • EMBOSS (livre) • http://emboss.sourceforge.net/ • Staden (livre) • http://staden.sourceforge.net/