Download
1 / 19
230 likes | 534 Views
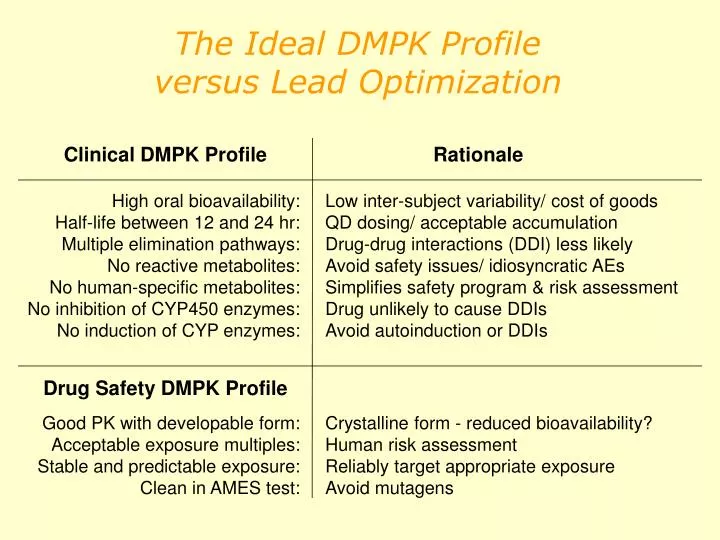
Clinical DMPK Profile. Rationale. High oral bioavailability: Half-life between 12 and 24 hr: Multiple elimination pathways: No reactive metabolites: No human-specific metabolites: No inhibition of CYP450 enzymes: No induction of CYP enzymes:. Low inter-subject variability/ cost of goods

E N D
Clinical DMPK Profile Rationale High oral bioavailability: Half-life between 12 and 24 hr: Multiple elimination pathways: No reactive metabolites: No human-specific metabolites: No inhibition of CYP450 enzymes: No induction of CYP enzymes: Low inter-subject variability/ cost of goods QD dosing/ acceptable accumulation Drug-drug interactions (DDI) less likely Avoid safety issues/ idiosyncratic AEs Simplifies safety program & risk assessment Drug unlikely to cause DDIs Avoid autoinduction or DDIs Drug Safety DMPK Profile Good PK with developable form: Acceptable exposure multiples: Stable and predictable exposure: Clean in AMES test: Crystalline form - reduced bioavailability? Human risk assessment Reliably target appropriate exposure Avoid mutagens The Ideal DMPK Profile versus Lead Optimization
Discovery DMPK In Vitro Assays Assay Rationale CYP 450 Inhibition: CYP 450 Profiling: Rat liver slice - induction: hPXR : Hepatocyte clearance: Inter-species hepatocyte met ID : Plasma stability: Plasma protein binding: Mock hERG drug assay: Caco-2: Avoid drug-drug interactions (DDI)? Avoid Polymorphism? Avoid DDI? Predict in vivo rat enzyme induction. Predict CYP 3A4 induction in humans. Predict human metabolic rate. Look for human-specific metabolites. Ex vivo degradation? Normalize exposure based on ‘free’ drug Confirm actual concentration. Predict human absorption potential.
Study Rationale Predict human PK Full” PK (IV & PO): Adequate exposure for safety testing Single-rising dose: Steady-state; tumorigenic induction 14-Day Rat multiple dose: Absorption and circulating metabolites ‘Hot’ PK (3H-SCH) : Identify metabolites in ‘safety’ species Metabolite Pathway Elucidation: Avoid unusual retention of metabolites Mass Balance of 3H-SCH: Melanin Binding (LE rats): Assess phototoxicity potential PK/PD: Estimate efficacious drug exposure Developable form acceptability: Adequate exposure for safety testing Discovery DMPK In Vivo Studies
Pharmacodynamics (PD) Effect vs Concentration Pharmacokinetics (PK) Concentration vs Time PK/PD Effect vs Time Pharmacokinetics and Pharmacodynamics Assumption: The magnitude of the desired effect (or side effect) is a function of the drug concentration at the site of action
Drug excretion (Metabolism-Elimination) Drug at Effect Site Drug at Absorption Site Drug in Tissues (Distribution) Response!! Drug at Effect Site Pharmacodynamics Pharmacokinetics PK-PD relationship PK What your body does to the drug PD What the drug does to your body
The test compound has a full effect at the dose of 10 mg/kg po Pharmacological effect Plasma level 16 cutoff 800 14 700 ng/ml 12 600 10 500 8 400 6 300 4 200 2 100 Veh 0 0 10 mpk, po 0 30 60 120 180 240 360 Time (min) At 10 mg/kg po the temporal profile of the plasma level and the pharmacological activity profile are similar The PK-PD relationship
Effect on the principal physiological system (cardiovascular, renal, nervous, etc). Safety Safety pharmacology Toxic effects could mechanism-based or compound-based. • To establish : • safe dose • MTD (maximum tolerated dose) • terapeutic window • target organs • Reversibility of toxicity Precinical toxicology Safety and preclinical toxicology
Preclinical toxicology • Acute toxicity profile • Chronic toxicity profile • 14 days toxicity test in one rodent and one non-rodent species before use in man. • 3 months study read out at 28 days • longer studies (12 & 24 month) • Mutagenicity tests in vitro and in vivo
Tolerability Therapeutic effect Sedation Ansiolitic effect What a therapeutic window is? Toxicity Respiratory depression
It is possible to obtain a clear separation between antithrombotic and bleeding effects P2Y12 antagonists Therapeutic index evaluation (van Giezen and HumphriesSemin Thrombosis Hemost2005)
Moving from Animals to Man • Humans and animals have different biochemistry, physiology and anatomy • Predictions of a drug’s PK profile in humans using animal PK data must account for these differences • Allometric scaling is used to predict differences based only on size. • The relationship of some PK parameters across species can be correlated with body weight. • One can determine an empirical relationship log PK parameters and log Body Weight • These parameters can be used to extrapolate PK parameters in humans when parameters have been determined in lower species (mouse, rat, dog, monkey, etc.) • The relationship is not always predictive, but it can often give a good estimate.
Summary The information collected in ADME and DMPK study are necessary to establish the dose that will be used in human
Pre-marketing Registration R&D process for a new drug CANDIDATE POC DRUG Exploratory development Full development Developpability Safety Therapeutic efficacy Fase 0 or Preclinical development Fase II Study in the patient Fase III Study in the patient Fase I (A and B) Fase IV Post marketing Surveillance
Formulation study • Pharmaceutical form: tablet , capsule, cream, injection, etc • To achieve the best effect is necessary to identify not only the • best form but also the most suitable formulation. • Example of composition of a tablet: • Active principle, filler, binder, • lubricant, disintegrant, surfactant.
Objectives of Clinical Trials • Phase I: First in man safety e tolerability • Phase II: First in patient IIa safety and tolerability IIb dose, dosage form • Phase III: Value (is better than existing treatments) • Post marketing surveillance or Phase IV : Monitor the drug in the real clinical setting
Clinical trials Uncontrolled Controlled Randomized Open or blind Sequential or cross-over






















