Download
1 / 36
360 likes | 382 Views
Explore the journey of inferring phylogenies, ranging from LUCA to modern species. Discover the evolving perspectives on phylogenetic trees and the utilization of molecular data and advanced algorithms. Dive into the intricacies of phylogenetic trees, defining features, and challenges in constructing accurate representations. Join us in unraveling the fascinating Tree of Life!

E N D
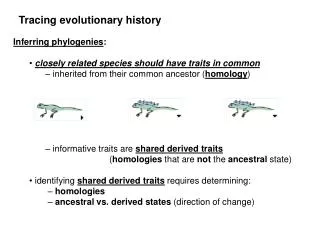
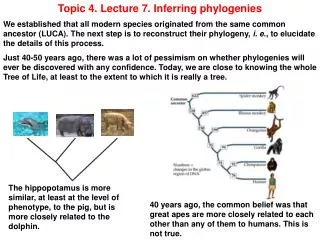
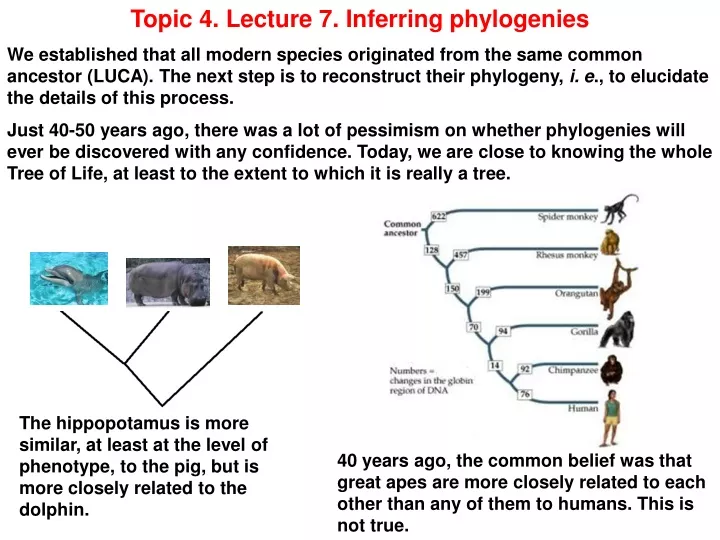
Topic 4. Lecture 7. Inferring phylogenies We established that all modern species originated from the same common ancestor (LUCA). The next step is to reconstruct their phylogeny, i. e., to elucidate the details of this process. Just 40-50 years ago, there was a lot of pessimism on whether phylogenies will ever be discovered with any confidence. Today, we are close to knowing the whole Tree of Life, at least to the extent to which it is really a tree. The hippopotamus is more similar, at least at the level of phenotype, to the pig, but is more closely related to the dolphin. 40 years ago, the common belief was that great apes are more closely related to each other than any of them to humans. This is not true.
This work has been performed at our Department! Land plants originated from green algae Charophyta, and relationships between their major clades have also been resolved.
There are two complementary reasons for progress in inferring phylogenies: 1) molecular data and 2) sophisticated algorithms and software tools. Indeed, a genome contains more traits than any higher-level description of the organism. Also, one does not need to be a specialist in a particular group of organisms. Every segment of genomes contains a lot of phylogenetic information: MSSHKTFRIKRFLAKKQKQNRPIPQWIRMKTGNKIRYNSKRRHWRRTKLG vertebrate Homo sapiens MASHKTFRIKRFLAKKQKQNRPIPQWIRMKTGNKIRYNSKRRHWRRTKLG vertebrate Danio rerio MPSHKTFRVKQKLAKAQKQNRPIPQWIRLRTGNTIRYNAKRRHWRKTRLG fungus Neurospora crassa MPSHKTFRTKQKLAKAQKQNRPIPQWIRLRTGNTIRYNAKRRHWRKTRLG fungus Podospora anserina MPSHKTFRIKQKLAKKQRQNRPIPYWIRMRTDNTIRYNAKRRHWRRTKLG plant Triticum aestivum MPSHKSFMIKKKLGKKMRQNRPIPHWIRLRTDNTIRYNAKRRHWRRTKLG plant Arabidopsis thaliana Alignment of amino acid sequences of ribosomal protein L39 from 6 species. Red - informative trait states (shared within vertebrates, fungi, or plants) Green - uninformative trait states (unique to a species) Blue and Yellow - misleading trait states (shared by distant species, due to homoplasy). To extract phylogenetic information from may of traits of many species, sophisticated algorithms and powerful computers are necessary. However, the basic ideas are simple.
The first question is: how can the phylogeny of a set of modern species be described? Such descriptions are provided by phylogenetic trees. A phylogeny can be described by a tree only if two key conditions are satisfied: i) variation within each lineage can be ignored, and each cladogenesis was an instant split, ii) there were no secondary merging of lineages. Violations of condition i) may become important only if we study very similar species. Violations of condition ii) are very important for prokaryotes, where lateral gene transfer is so common that we often encounter "phylogenetic networks", instead of trees. Mathematically, a tree is a graph without cycles.
Terminology: The lineage which corresponds to the common ancestor is called the ROOT of the tree; extant species (and, perhaps, extinct species which left no descendants) are LEAVES; and intermediate ancestors are INTERNAL NODES; all these are connected by EDGES. Two most closely related species are SISTERS, any other is an OUTGROUP to them. Complete branch = clade
Suppose that a phylogeny we study can, indeed, be represented by a tree. A phylogenetic tree has two properties: i) Each leaf carries a unique label - a leaf corresponds to a particular species! ii) The tree is binary: each cladogenesis produces only two branches. A trifurcation simply means that we cannot determine the order of bifurcations.
A phylogenetic tree describes how the common ancestral lineage branched off to, eventually, form the set of extant species. The order in which different lineages split is called topology of the tree, and topology is more fundamental than, say, the relative lengths of different branches of the tree. Top: 4 trees with the same topology. Bottom: a tree with a different topology.
So, we have a set of N species that evolved from the common ancestor, and want to discover their phylogenetic tree. How do we proceed? A naive idea is to try all possible "substantially different" trees and to chose the one which fits the data best. However, with N > 10-15, trying all possible trees is impossible. Discovering the phylogeny by trying all possible trees is an "NP-hard" (non-polynomially hard) problem - because their number T(N) increases exponentially with N. Obviously, there is just one possible tree for 2 species, so that T(2) = 1. We can attach the 3rd species to this tree is 3 ways, so that T(3) = 1 x 3.
To each of the 3 possible 3-species trees, we can attach the 4th species in 5 ways, so that T(4) = 1 x 3 x 5 = 15. Five 4-species trees that can be built from the 3-species tree #1. T(4) = 15.
Thus, T(N) = 1 x 3 x 5 x 7 x ... x (2n-3) = {1 x 2 x 3 x 4 x ... x (2n-3)}/{2 x 4 x ... x (2n-4)} = = (2n-3)!/2n(n-2)! When N increases, T(N) increases faster than any polynomial in N - that is why considering all possible trees is an "NP-hard" problem. Number of species N Number of trees T(N) 1 1 2 1 3 3 4 15 5 105 6 945 7 10,395 8 135,135 9 2,027,025 10 34,459,425 20 8 x 1021 30 5 x 1038 40 1 x 1057 50 3 x 1076
In addition to cladogeneses, a tree can also display changes of the trait states. A phylogenetic tree based on a segment of a protein from Homo sapiens, Mus musculus, and Rattus norvegicus. A phenotype consists of 6 traits, each associated with a position within the alignment. Actual phenotypes of the 3 extant species and the inferred phenotypes of the common ancestor of mouse and rat and of the whole set are shown, together with evolutionary events. Traits 1 and 6 are invariant and, thus, of no help.
So, we cannot simply try all possible trees and see which one of them fits the data best (even if we know how to evaluate this fit). What can we do instead? Let us start from two simple cases when reconstructing a phylogeny is easy: I) Purely divergent evolution, without any homoplasy - as revealed by lack of pairwise conflicts in the matrix of traits (hierarchical joint distribution). II) Constant-rate evolution (or evolutionary clock) - as revealed be identical distances from any two sisters to the outgroup. DIFFICULT MATERIAL FROM HERE! I) Matrix of traits, consistent with purely divergent evolution II) Matrix of distances, consistent with constant-rate evolution
Special case I. Reconstructing divergent evolution. Theorem. If a set of species originated from the common ancestor by (exclusively) divergent evolution, the phylogenetic tree which describes this evolution is maximally parsimonious (i. e., involves the minimal possible number of changes). Proof. Obvious - a tree which involves more than the minimal number of changes must involve some homoplasy (repetitive origins of the same trait state). OK, but how do we know if evolution of our set of species was divergent? We already know the answer: if the joint distribution of traits contains pairwise conflicts between traits, evolution must involve homoplasy. In contrast, if this distribution is hierarchical, the hypothesis of exclusively divergent evolution is not rejected - and is probably true. Without homoplasy, a derived trait state always define a clade. Synapomorphy is a fancy word for such a shared derived trait state. Conflict between two traits.
We need only to make one last step - to produce the maximally parsimonious tree from a hierarchical matrix of traits. There is a simple algorithm for this (traits are binary): 1) Choose a trait with the minimal number of species having the derived state (if more that one trait has this number, choose any one). Take all species which possess the derive state - they form the lowest-level clade. 2) Forget about the trait just used, and repeat this procedure, until all the traits are used. Let us see how this works:
Venn diagram representing the same data. For each trait, species with the trait state shown in red in the table are enclosed into a line of the corresponding color. Let assume that these states are derived. Trait 116 in involved in two conflicts - we will ignore it.
Let us remove trait 116. After this, the algorithm can be applied. This is one of several maximally parsimonious trees that are consistent with our matrix of traits (minus trait 116). Find other possibel MP trees. If several different trees are equally parsimonious, each of them can be used - we cannot know which one is correct.
A phylogenetic tree reconstructed using the above matrix of traits, ignoring trait 116. Only the phenotype of the last common ancestor is shown, but phenotypes of all the intermediate ancestors have also being reconstructed.
Above, we assumed that we know, for each trait, which state is ancestral (or primitive, or plesiomorphic) and which is derived (apomorphic). For this, several methods can be used: 1) Rely on partial a priori understanding of evolution: for example, vestigial eyes or an inserted pseudogene or a TE must be derived states (why?). 2) Rely on fossils: trait states that appear later in the fossil record (e. g., wings of birds), must be derived.
3) Rely on an outgroup - one or several species that definitely branched off before the series of cladogeneses which we want to reconstruct. How do we know that something is an outgroup? By assuming approximately constant rate of evolution - mice are certainly an outgroup for humans and chimpanzees. Then, for a trait that is variable within the set of species we study, its state present in the outgroup is probably ancestral, as long as the outgroup is not too far away. Mus must be an outgroup to hominids! Drosophila is also an outgroup to hominids, but much less useful. So, an outgroup must be not too close but also not too distant.
4) A more fancy approach: first, construct an unrooted tree, without knowing which traits states are derived (this is only slightly more complex than what we did), and then root the tree. The number of unrooted trees U(N) = T(N-1) - still a lot for large N, but unrooted phylogenies start from N = 4. There is only one unrooted tree of 3 species, and 3 unrooted trees of 4 species.Of course, an unrooted tree does not contain full information about past evolution. One possibility is to assume near-constant rate of evolution, and root the tree in the middle.
OK, but what to do if we there are many conflicts between traits? If all conflicts are due to a small number of "bad" traits, these traits can be ignored, and hierarchy restored. However, multiple conflicts prove that evolution of the set of traits was significantly non-divergent. In this case the maximally parsimonious tree is not a solution, because: a) this tree is hard to find - generally, this is an NP-hard problem. b) this tree may not correspond to reality - why to expect minimal homoplasy? Here a different approach is worth trying, which works in the other special case. Special case II. Reconstructing constant-rate evolution. Suppose that we have data on a large enough number of traits, and that evolution of our set of species occurred at a constant rate (evolutionary clock). If so, the distance between phenotypes of two species immediately tells us how long ago their lineages split. Thus, a symmetric NxN matrix of distances between our N species can be immediately converted it into their phylogenetic tree.
Let is convert the above matrix of traits into the symmetrical matrix of distances between the species. The distance is the number of traits in different states. Here, the number of traits is too small to assume constant-rate evolution.
A more suitable example: matrix of distances between second introns of beta actin gene from human Homo sapiens, chimpanzee Pan troglodytes, orangutan Pongo pygmaeus, rhesus macaque Macaca mulatta and common marmoset Callithrix jacchus. The phylogenetic tree can be recovered from the matrix of distances using an algorithm UPGMA (Unweighted Pair Group Method with Arithmetic mean): 1) find the most similar pair of species; 2) replace these two species with a clade consisting them. The distances to this clade are arithmetic means of distances to the two removed species. 3) repeat this procedure. The distance matrix after the first step of UPGMA. Naturally, N-1 steps are needed to construct the tree, rooted at the middle. Here similarity = relatedness.
However, appliyng UPGMA when evolution was not constant-rate can lead to errors. Matrix of distances between human Homo sapiens, dog Canis familiaris, and mouse Mus musculus. Here, the distance is the estimated per site number of synonymous nucleotide substitutions. A wrong tree obtained from this matrix by UPGMA (left) and the true tree (right).
Analogously to Case I, a simple test can tell whether our data are consistent with the assumption of Case II. The evolutionary clock hypothesis implies that distances AC and BC must be equal (where C is a definite outgroup for sisters A and B). This is a powerful test! Evolutionary clock possible Evolutionary clock impossible
Often, data FAIL both tests - there are conflicts, and distances from sister species to the outgroup are not the same - revealing that evolution was not exclusively divergent and did not proceed at an exactly constant rate. What to do? More sophisticated approaches can be used. Maximal likelihood - chose the hypothesis H (tree) which generates the data D (matrix of traits) with the highest probability Prob(D|H). This quantity is also known as the likelihood of the hypothesis, given the data. However, to calculate Prob(D|H) we need to have some idea of how evolution occurred - for example, that some traits evolve faster than others. Bayesian methods – extension of the previous approach, under assumption that we have some a priori idea of what trees are more probable. Things get technical at this point. However, the best solution is to find better traits, variable enough to convey some information on evolution, but still not producing homoplasy often. Such traits do exist, for each depth of phylogeny. Homoplasy is the worst enemy of phylogenetic reconstruction!
How to find good traits for phylogenetic reconstructions? 1) Phylogenies of very similar species can be based on substitutions at individual sites of junk DNA. 2) Phylogenies of moderately similar species canbe based on short inversions: AAACGAGGGAAA > AAACCCTCGAAA Indeed, two independent inversions will rarely have the same boundaries. Inspect pairs of columns, and look for all 4 possible combinations of red and green - are there any conflicts? 3) For yet more distant species, traits associated with gene orders can be used. 4) For very distant species, only tratis associated with sequences of conservative proteins can be used, and in this case homoplasy cannot be ruled out.
OK, but are phylogenies trees? Non-zero width of edges should sometimes be taken into account, because it can lead to "lineage sorting", but this is not a big deal at the Macroevolutionary scale. Even in eukaryotes occasional episodes of lateral gene transfer (LGT) are well-documented. Still, eukaryotic phylogenies are generally tree-like. In bacteria, LGT is pervasive, although there still is a tree-like "phylogenetic signal".
Why do we care to know phylogenies? First, because, this is something we want to know! Second, hard-to-discover phylogenetic affinities can be really important.
Comparing phylogenies can reveal patterns in coevolution. Phylogenies of primates (left) and their malaria parasites (right).
Phylogenies are important not only in biology. Ohylogeny of Indo-European languages; numbers indicate inferred times of divergence.
Thoughts about classification (you can safely forget them) Any classification somehow reflects similarity between the objects we classify. However, not all classifications are hierarchical. For example, the periodic table of elements is not hierarchical, and does not reflect common ancestry. This is because chemical properties of atoms can change radically after a single event: 40K decays into 40Ar.
In contrast, classification of species (at least of eukaryotes) is hierarchical, because evolution is slow and gradual, and common ancestry strongly affects similarity. The only contentious issue is whether it is OK to use paraphyletic taxa. Can we say dinosaurs, or only non-avian dinosaurs (when we want to exclude birds)? monophyly (holophyly) polyphyly paraphyly My answer to is "who cares"? A classification cannot be true or false - it can only conform (or not conform) to some set of rules. In contrast, a phylogeny can be true or false. The only real issue is to discover the true phylogeny - and how we describe it using the language of classification is not important.
Conclusions about phylogenetic reconstructions: • In eukaryotes phylogenies are mostly trees, in prokaryotes LGT is very common. • 2) In simple cases we can easily find the correct tree, assuming either exclusively divergent or constant-rate evolution. • 3) If neither of these assumptions is consistent with the data, we can either use more sophisticated methods or, preferrably, seek better traits, not prone to homoplasy. • 4) A lot of phylogenetic relationships have already being elucidated, and the whole Tree of Life will be known soon.
Quiz: consider this matrix of traits: Is this matrix consistent with exclusively divergent evolution? If not, home many traits are responsible for homoplasy? Construct the phylogenetic tree, assuming that 1 is always a derived state.