Download
1 / 33
340 likes | 833 Views
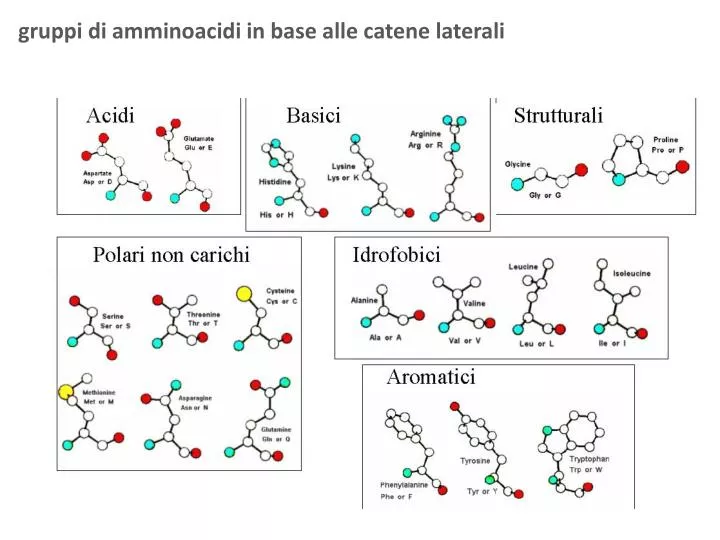
gruppi di amminoacidi in base alle catene laterali. STRUTTURE TERZIARIE (singola catena polipeptidica ). Proteine multi-dominio. Un unico dominio strutturale. STRUTTURE QUATERNARIE (associazioni di diverse catene polipeptidiche ).

E N D
STRUTTURE TERZIARIE (singola catena polipeptidica) Proteine multi-dominio Un unico dominio strutturale STRUTTURE QUATERNARIE (associazioni di diverse catene polipeptidiche)
UniProt raccoglie le informazioni dei database Swiss-prot e TrEMBL . Viene curato anche un database NON RIDONDANTE (UniRef). Banche dati di sequenze proteiche Molto curato e dettagliato, con annotazioni circa funzione, struttura, modificazioni e altre informazioni utili E’ la traduzione in silicodi ogni entry codificante del database nucleotidico dell’EMBL, non è accurato, ma è ricchissimo
Fast-A FORMAT >tr|P73799 Slr1259 protein - Synechocystis sp. (strain PCC 6803). MLFRQLFDPETSTYTYVIADPKGRSAALVDSVLEQVDRDLNLLKELDLKLTFCLETHVHADHITGAGKLRQLTGCQNLVPQYAEVDCADRHLQDGEIVHVGSIPIQAIATPGHTDSHLAFLVNQTHVLTGDALLIRGCGRTDFQSGDAGTLYDAIHGKLFTLPEDVFVYPGHDYRGHTVSTIGEEKRFNPRLLGRDRQNFIEFMDSLNLPDPKKIMEAVPANQLCGQRTVAV
Avendo l’abbonamento si ottiene l’intero articolo • (formato pdf) • Consultazione banche dati per cercare • gli articoli Banche dati di letteratura www.pubmed.org PubMed/Medline www.biblio.unimib.it Biblioteca d’Ateneo http://apps.isiknowledge.com/ ISIWeb of Knowledge
ALLINEAMENTO DI SEQUENZE A COPPIE AGTTTGAATGTTTTGTGTGAAAGGAGTATACCATGAGATGAGATGACCACCAATCATTTC ||||||||||||||||||| |||||||| ||| | |||||| ||||||||||||||||| AGTTTGAATGTTTTGTGTGTGAGGAGTATTCCAAGGGATGAGTTGACCACCAATCATTTC MULTIPLO KFKHHLKEHLRIHSGEKPFECPNCKKRFSHSGSYSSHMSSKKCISLILVNGRNRALLKTl KYKHHLKEHLRIHSGEKPYECPNCKKRFSHSGSYSSHISSKKCIGLISVNGRMRNNIKT- KFKHHLKEHVRIHSGEKPFGCDNCGKRFSHSGSFSSHMTSKKCISMGLKLNNNRALLKRl KFKHHLKEHIRIHSGEKPFECQQCHKRFSHSGSYSSHMSSKKCV---------------- KYKHHLKEHLRIHSGEKPYECPNCKKRFSHSGSYSSHISSKKCISLIPVNGRPRTGLKTs
Allineamento GLOBALE o LOCALE GLOBALE considera la similarita’ tra due sequenze in tutta la loro lunghezza (da N- a C-terminale) LOCALE considera solo specifiche REGIONI simili tra alcune parti delle sequenze in analisi (solo regioni a ↑ densità di similarità generando più sub-allineamenti) Global alignment LTGARDWEDIPLWTDWDIEQESDFKTRAFGTANCHK ||. | | | .| .| |||| | || TGIPLWTDWDLEQESDNSCNTDHYTREWGTMNAHKAG Localalignment LTGARDWEDIPLWTDWDIEQESDFKTRAFGTANCHK ||||||||.|||| TGIPLWTDWDLEQESDNSCNTDHYTREWGTMNAHK
ALLINEAMENTO DI SEQUENZA • PER ESEGUIRE UN ALLINEAMENTO DI SEQUENZA SONO NECESSARI ESSENZIALMENTE 3 STRUMENTI: • Avere a disposizione unaMATRICE DI SOSTITUZIONE. La matrice definisce la il GRADO di SIMILARITA’ tra amminoacidi; • Avere a disposizione unALGORITMO DI ALLINEAMENTO cercando di massimizzare il punteggio dato dalla matrice e valutando quanti gap (interruzioni) inserire; • Avere a disposizione per evitare allineamenti senza senso una PENALITA’ per l’introduzione dei GAP. I GAP riflettono inserzioni/delezioni avvenute durante l’evoluzione LLTTVRNN LLTTVRNN LLVRNN LL--VRNN
Similarità e distanza Esistono due modi per misurare il grado di omologia tra due sequenze: • Calcolare la similarità contando i match • Calcolare la distanza contando mismatche indels Similarità elevata ↔ bassa distanza Due sequenze identiche hanno una distanza pari a zero
SIMILARITA’ DI SEQUENZA • Nel punteggio di similarità di sequenza si tiene conto del fatto che gli amminoacidi a confronto in ogni posizione siano simili, differenti o identici e di una penalità per i gap. • PER DEFINIRE LA SIMILARITA’ TRA LE DUE SEQUENZE SI USANO MATRICI BASATE SU PRESUPPOSTI DIVERSI: • identità/non identità; • Caratteristiche chimico-fisiche degli aa; • Basate sul codice genetico: valutare quante mutazioni fare in una tripletta per passare da un aa a un altro. (se ad es. si cambia un solo nucleotide la sostituzione la sostituzione sarà meno penalizzata perché si tratta di evento probabile nel corso dell’evoluzione) • Basate sucriteri evolutiviestrapolati da confronto di sequenze di proteine omologhe (MATRICI BLOSUM E PAM) 2 penalità per i gap (apertura (fisso), estensione (lunghezza dipendente))
Quale matrice PAM conviene utilizzare? • In generale per due sequenze filogeneticamente vicine è meglio utilizzare una matrice PAM a basso indice e viceversa • In assenza di informazioni si utilizzano PAM40, PAM120 e PAM 250 • PAM250 individua similarità del 20% • PAM120 individua similarità del 40% • PAM80 individua similarità del 50% • PAM60 individua similarità del 60%
L’utilizzo della matrice di similarita’ appropriata per ciascuna analisi e’ cruciale per avere buoni risultati.Infatti relazioni importanti da un punto di vista biologico possono essere indicate da anche molto debole similarità. Sequenze poco divergenti molto divergenti BLOSUM80 BLOSUM62BLOSUM45 PAM1 PAM120PAM250
ALLINEAMENTI MULTIPLI L’allineamento multiplo è un’ipotesi di omologia posizionale tra basi o aminoacidi Tutti i residui presenti nella stessa colonna di un multi-allineamento sono evolutivamente correlati No applicabili algoritmi di allineamento globale esatto (cresce esponenzialmente con il numero di sequenze da allineare) Teoricamente sarebbe possibile applicare l’algoritmo di allineamento globale, ma in pratica non lo è perché richiede tempi di esecuzione troppo lunghi METODI APPROSSIMATI Es. ALLINEAMENTO PROGRESSIVO (implementato in Clustal W)
ALLINEAMENTO PROGRESSIVO • Basato su costruzione di una successione di allineamenti a coppie • Dato un insieme S costituito da n sequenze da allineare, si scelgono due sequenze s1 e s2 e si allineano; questo allineamento resta fissato nei passi successivi • Si sceglie una terza sequenza s3 e si allinea al precedente allineamento, e così via • Prevedono che coppie di sequenze che presentano un maggior grado di similarità tra loro siano allineate per prime (giustificato dal fatto che coppie di seq + simili avranno maggiore probabilità di essere derivate + recentemente da un antenato comune e il loro allineamento fornisce informazioni più affidabili - le posizioni dei gap in seq maggiormente correlate sono tipicamente + accurate rispetto a quelle relative a seq meno simili, per cui i gap degli allineamenti iniziali vanno preservati durante l’allineamento progressivo)
A B D C ClustalW • Otterremo un albero i cui rami hanno lunghezza proporzionale alla distanza tra le sequenze: • Quest’albero verrà utilizzato per guidare l’allineamento progressivo. • Nel nostro esempio verranno allineate per prime le sequenze A e B. Successivamente verrà allineata la sequenza D all’allineamento AB e infine verrà allineata la sequenza C all’allineamento ABD.
Allineamento con ClustalW • La presenza di un simbolo * in fondo ad una colonna indica un match del 100%. • Il simbolo :indica un’alta similarità (>75%). • Il simbolo.indica una media similarità (50%-75%).
INPUT CLUSTALW Sito EBI FORMATO RICHIESTO: FastA
OUTPUT CLUSTALW Sito EBI Tabella riassuntiva con link a: file di OUTPUT (con le indicazioni sulla costruzione dei gruppi) - allineamento
File di output Elenco seq – attribuzione di un numero a ogni seq Confronti a coppie Formazione gruppi
Quando le sequenze da allineare non sono molto divergenti (similarita’>45% per le proteine) ClustalWdàunarispostaottimale • In casocontrariosononecessariaggiustamenti (correzionemanualedell’allineamento) • Unavoltache 2 o + seqsiano state allineate in un blocco, questoallineamento è fissato e non puòpiùesseremodificatonellefasi successive dell’allineamentoprogressivo. • Problemidiminimo locale: se in unaqualunquefasevieneintrodotto un erroresipropagheràsullefasi successive • Affidabilità del multiallineamentodipendedanchedal set diseq considerate – es. Se si include unaseq non realmenteomologaallineamentoprodottorisultaalteratodainserzionemolti gap addizionali (controlloseq input per rimuoverequelle “spurie”)
Una sequenza “da sola” non e’ informativa, è utile poterla confontare alle sequenze note nei database perche’ possano essere formulate delle ipotesi sulla sue relazioni evolutive con sequenze simili o sulla sua funzione. • Metodi di ricerca di similarità in banca dati: programmi che permettono di fare lo “screening” di una banca dati usando una sequenza “sonda”/”esca” (detta query) come input ( le sequenze nel DB sono chiamate subject) • Devono essere veloci, selettivi e sensibili • Si basano su metodi euristici • Utilizzano allineamentilocali per confrontare • le sequenze • Algoritmo “Euristico” = in matematica e informatica un particolare tipo di algoritmo la cui soluzione non è la soluzione ottima per quel dato problema ma una soluzione approssimativamente molto vicina a quella ottima con tempi di calcolo ragionevoli. RICERCA DI SIMILARITA’ IN BANCHE DATI
Valutazione significatività dei match identificati Quanto il match (query vs seq x del DB) identificato dagli allineamenti locali di BLAST è significativo? Tanto più il loro allineamento è diverso da uno generato casualmente tra sequenze di lunghezza paragonabile Sequenze che danno un allineamento casuale: –Sequenze rimescolate(“shuffled”) –Sequenze generate casualmente N.B. Blast permette di mascherare le regioni di sequenza a bassa complessitè
E-value E-value= expectation value, numero atteso di sequenze che danno per caso il punteggio opt Indica quanto e’ probabile che si trovi il punteggio S per caso in una distribuzione di Poisson con valore medio Mcasuale NB IN BLAST il punteggio OPT puo’ essere convertito in scala logaritmica al punteggio cosidetto BIT Indicazioni: opt/bit elevati, Evalue prossimo a 0
BLAST blastp cerca in database di sequenze proteiche usando come query sequenze proteiche blastn cerca in un database di sequenze nucleotidiche usando come query sequenze nucleotidiche blastx cerca in un database di sequenze proteiche partendo da una sequenza query nucleotidica che viene tradotta in tutti i frame tblastn cerca in un database di sequenze nucleotidiche partendo da una sequenza query proteica – le seq subject del database sono tradotte in sequenze proteiche in tutti i frame PSI-Blast ricerca iterativa con PSI-Blast usando ad ogni iterazione una sequenza consenso derivata dall’allineamento tra la seq query le le subject dell’interazione precedente
Le proteine ed i domini proteici che appartengono ad una particolare famiglia generalmente condividono attributi funzionali e derivano da un “antenato” comune. Dallo studio di sequenze risulta evidente che alcune regioni si conservano meglio di altre nel corso dell’evoluzione. Queste regioni in genere sono importanti per il mantenimento della struttura tridimensionale o per la funzione di una proteina. Analizzando le proprietà che vengono mantenute costanti e quelle che invece variano è possibile ottenere una “signature” per ogni famiglia proteica o dominio che consente di distinguere i suoi membri dalle altre proteine non correlate.
Ricerca di pattern e motivi funzionali Uno dei primi scopi della biologia computazionale consiste nel rispondere alla domanda: data una nuova sequenza, cosa si può dire sulla funzione (o funzioni) in essa codificata? Se la ricerca per similarità non fornisce risposte si devono usare altri strumenti: la ricerca di pattern e motivi funzionali La pattern recognitionè un’area di ricerca dell’informatica e della fisica applicata con ricadute in campi quali l’intelligenza artificiale, la linguistica computazionale, le scienze cognitive, la statistica matematica, ecc. Studia l’organizzazione e il disegno di sistemi che riconoscano motivi e regolarità nei dati a disposizione
Un motivo (pattern) è un insieme di caratteri (nucleotidi o aminoacidi), non necessariamente contigui, associati spesso ad una precisa struttura o funzione La loro esistenza dipende dal fatto che l’evoluzione ha prodotto pochi modi per realizzare una determinata funzione Motivo ideale può essere sempre ed univocamente associato ad una precisa struttura o funzione Motivo reale si trova in sequenze che non presentano la funzione (falsi positivi) o è assente in sequenze funzionalmente correlate al motivo (falsi negativi) I motivi possono essere codificati in diversi modi
BANCHE DATI DI MOTIVI • All’interno di un singolo motivo l’informazione può essere ridotta a una SEQUENZA CONSENSO che non deve essere necessariamente stringente: PATTERN; • Se ci si riferisce a un gruppo di motivi conservati non contigui nella sequenza: FINGERPRINT oppure BLOCCHI; • Se invece non si identificano regioni locali di similarità tra proteine di una stessa famiglia ma l’informazione viene dal considerare la similarità lungo l’intero allineamento si ha un PROFILO
MOTIVI…E MOTIVI PATTERN FINGERPRINT O BLOCCHI PROFILO : possibilità di ricavare una sequenza consenso per tutto l’allineamento XXXXhhhhXXXbbxxaaxxNGG(X)5-8SWXX…
Ricerca di pattern e motivi funzionali in sequenze proteiche Dallo studio di allineamenti multipli di sequenze appartenenti ad una stessa famiglia di proteine, appare evidente che alcune regioni sono più conservate regioni importanti per la funzione o la struttura Le regioni più conservate sono in genere quelle più importanti per la funzione Dalle regioni costanti e variabili di un multi-allineamento di proteine omologhe derivare un pattern che serva a distinguerle, cioè si può identificare un motivo che possa servire alla caratterizzazione funzionale delle proteine che lo contengono. Esistono diversi programmi per l’individuazione di motivi: PROSITE (patterns, profili e patterns a alta probabilita’ di accadimento) classificazione funzionale della proteina e predizione putative modifiche post-traduzionali PFAM (profili) classificazione di domini strutturali