Download
1 / 48
560 likes | 978 Views
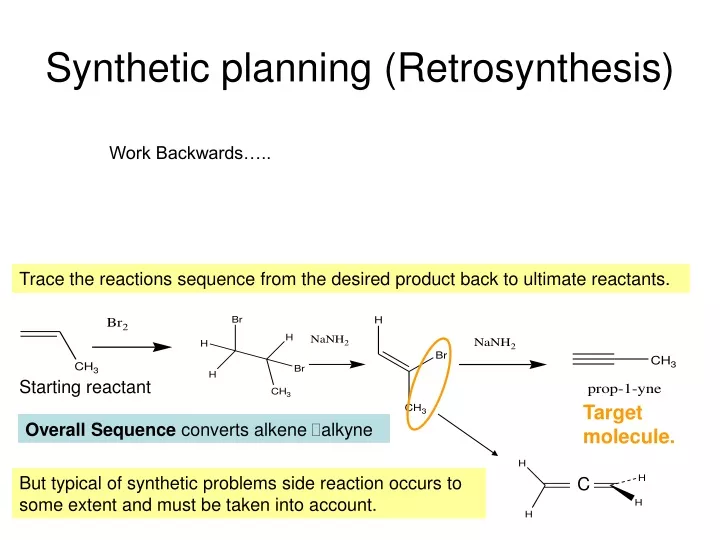
Synthetic planning (Retrosynthesis). Work Backwards…. Trace the reactions sequence from the desired product back to ultimate reactants. Starting reactant. Target molecule. Overall Sequence converts alkene alkyne.

E N D
Synthetic planning (Retrosynthesis) Work Backwards….. Trace the reactions sequence from the desired product back to ultimate reactants. Starting reactant Target molecule. Overall Sequence converts alkene alkyne But typical of synthetic problems side reaction occurs to some extent and must be taken into account. C
More Sythesis: Nucleophilic Substitution Use the acidity of a terminal alkyne to create a nucleophile which then initiates a substitution reaction. Note that we still have an acidic hydrogen and, thus, can react with another alkyl group in this way to make RCCR’ Alkyl halides can be obtained from alcohols
Reactions: alkyne with halogen RCCR + Br2 RBrC=CBrR No regioselectivity with Br2. Stereoselective for trans addition.
Reactions: Addition of HX The expected reaction sequence occurs, formation of the more stable carbocation. Markovnikov orientation for both additions. Now for the mechanism….
Mechanism The expected reaction sequence occurs, formation of the more stable carbocation.
Reactions: Acid catalyzed Hydration (Markovnikov). Markovnikov addition, followed by tautomerism to yield, usually, a carbonyl compound.
Reactions: Anti Markovnikov Hydration of Alkynes, Regioselectivity Step 2 Step 1 Similar to formation of an anti-Markovnikov alcohol from an alkene Step 1, Internal Alkyne: addition to the alkyne with little or no regioselectivity issue. Alternatively Asymmetric, terminal, alkyne if you want to have strong regioselectivity then use a borane with stronger selectivity for more open site of attack. Less exposed site. Aldehyde not ketone. More exposed site. sia2BH
Tautomerism, enol carbonyl Step 2, Reaction of the alkenyl borane with H2O2, NaOH would yield an enol. Enols are unstable and rearrange (tautomerize) to yield either an aldehyde or ketone. Overall… internal alkyne ketone (possibly a mixture, next slide) Terminal alkyne aldehyde
Examples Used to insure regioselectivity. As before, for a terminal alkyne. But for a non-terminal alkyne frequently will get two different ketones Get mixture of alkenyl boranes due to low regioselectivity.
Reduction, Alkyne Alkene 1. Catalytic Hydrogenation If you use catalysts which are also effective for alkene hydrogenation you will get alkane. You can use a reduced activity catalyst (Lindlar), Pd and Pb, which stops at the alkene. You obtain a cisalkene. Syn addition
Reduction - 2 2. Treatment of alkenyl borane with a carboxylic acid to yield cis alkene. Instead of H2O2 / NaOH Alkenyl borane 3. Reduction by sodium or lithium in liquid ammonia to yield the trans alkene.
Plan a Synthetic SequenceRetrosynthesis YES! Synthesize butan-1-ol from ethyne. Work backward from the target molecule. A big alkyne can be formed via nucleophilic substitution. This is the chance to make the C-C bond we need. Is read as “comes from”. Major problem: make big from small. Be alert for when the “disconnect” can be done. Catalytic Lindlar reduction • BH3 • H2O2, NaOH Convert ethyne to anion and react with EtBr. Do a “disconnect” here. Target molecule Catalytic reduction Lindlar Addition of HBr. Now, fill in the “forward reaction” details Can we get an alkyne from smaller molecules? Not yet! So how can we get it? How about joining molecules to get an alkene? Not yet!! So how can we get an alkene? Ask yourself! Do we know how to join any two molecules together to yield an alcohol?
Boiling Points The size of –Br and –CH3 about the same but bromo compounds boil higher due to greater polarizability; more dispersion forces.
Reaction of elemental halogen and alkanes yielding haloalkanes H3C – H + X – X H3C – X + H - X • Reaction Characteristics • Requires heat or light to initiate. • Fluorine is explosive. Reactivity decreases fluorine to chlorine to bromine to iodine which does not react. Bond Dissociation Energies. Energy Required to Break Bond. Chlorination: DH = 439 + 247 -351 – 431 = -96 kJ exothermic Bromination: DH = 439 +192 – 301 – 368 = -38 kJ less exothermic
Regioselectivity Starting Point in Analysis: Random Substitution. Assume that all hydrogens are equally likely to be replaced by X. There are 8 H in the molecule. Equally likely to be replaced if random….
Regioselectivity Now include experimental results for X = Cl, Br. • Replacement of secondary H is favored over primary H. Generally order of reactivity is tertiary > secondary > primary > methyl. • Bromination displays greater selectivity than does chlorination.
Reactivities of Hydrogens For chlorination tertiary:secondary:primary = 5:4:1 For bromination tertiary:secondary:primary = 1600:80:1 Quantitative Analysis: Deviations from random replacement quantified by assigning a Reactivity to each kind of Hydrogen (primary, secondary, or primary). Substitution at a carbon proportional to (# hydrogens at C) x (Reactivity of the H’s)
Predict product mixture For chlorination of 2,2,4,4-tetramethylpentane predict the product mixture. Cl2 + UV light 18 x 1 = 18 2 x 4 = 8 Number of hydrogens leading to this product Expected ratio 9 : 4 Expected fraction 9/13 4/13 Reactivity of those hydrogens
Chain Reaction Mechanism 1. Cl – Cl 2 Cl. The life of the chain depends on the ongoing presence of the highly reactive Cl atoms and alkyl radicals. Eliminating these species ends chains. 4. 2 Cl. Cl – Cl 5. 2 R. R – R 6. R.+ Cl. R - Cl Weak Cl-Cl bond may be broken by heat or light. Initiation Heat or light Chlorine atom. Highly reactive, only seven electrons in valence shell 2. R – H + Cl. R. + H - Cl Chain steps. Alkyl radical, only seven electrons around the C, highly reactive alkyl radical. 3. R. + Cl – Cl R – Cl + Cl. Repeat 2, 3, 2, 3,…. Hydrogen to be abstracted. Trade bonds: R-H for H-Cl Regenerates the Cl atom used in step 2 Termination steps. Trade bonds: weak Cl-Cl for a stronger C-Cl
Energetics of the Chain Steps Chlorination of ethane Step 2, abstraction of the H, controls the regioselectivityof the reaction. Isothermic or slightly exothermic for Cl; endothermic for Br. Step 3, attachment of the halogen, controls the stereochemistry, which side the halogen attaches. Exothermic
Step 2 and Bond Dissociation Energies, breaking bonds… More highly substituted radicals are easier to make. This gives rise to regioselectivity = non-random replacement.
Compare chlorination and bromination of 2-methyl propane. Bromination is more Regioselective. Examine Step 2 only for regioselectivity. First chlorination. Two kinds of H. BDE, reflecting different radical stabilities Slightly exothermic Now bromination. H-Cl is a more stable bond than H-Br. Step 2 is exothermic for chlorination. Endothermic for Br Endothermic
Step 2 Transition State Energetics, Cl vs Br Early transition states, little difference in energies of activation, rates of abstraction and regioselectivity chlorination R…H……….Cl Halogenation of 2-methylpropane yields two differerent radicals, primary and tertiary. Exothermic, tertiary radical more stable R……….H..Br Late transition states, larger difference in energies of activation, rates of abstraction and regioselectivity. bromination Endothermic, but tertiary radical still more stable by same amount.
Now Step 3: Stereochemistry Mirror objects. If a chiral carbon has been produced we get both configurations. Alkyl radical, sp2 hybridization, one electron in the p orbital. • Step 3 has these characteristics • Determines stereochemistry • Is exothermic • Is fast, not rate determining
Simple Example: monochlorination of 2-methylbutane From a and a’. a a’ b c Chiral carbon d From b First, look carefully at molecule Observations: Optically inactive molecule (can show reflection plane) and products will be optically inactive. From c Four optically inactive fractions if distilled. From d Chiral carbon Next, organize approach, label the carbons.
Example Can get relative amounts made of each using reactivities of 1:4:5 First get stereochemical relationships between carbons. enantiomers a’ a a a’ c b b’ 3 x 1 3 x 1 b enantiomers b’ b’ b enantiomers 1 x 5 / 2 Diastereomers both sides used. 1 x 5 / 2 1 x 5 / 2 Diastereomers both sides used. 1 x 5 / 2 c c Distillation would yield 5 optically inactive fractions. meso meso 2 x 4 / 2 2 x 4 / 2
Allylic Systems CH3CH2 – H: 411 kJ/mol (101 kCal/mol) Vinyl C – H bonds, difficult to break. Allylic C – H bonds, weak easily broken. Removal of H produces the allylic radical. Now the allylic radical…
Resonance in Allylic Radical Resonance provides the stabilization. The pi system is delocalized. Odd electron located on alternate carbons, C1 and C3, not C2.
Allylic Substitution Allylic C-H, 372 kJ H-Br 368 kJ Br-Br 192 kJ Allylic C-Br 247 kJ DH = -247 - 386 – (-372 – 192) = -51kJ
Mechanism initiation Weakest C-H bond selected, highest reactivity Chain steps 368 kJ 372 kJ Termination: usual combining of radicals
We have a Problem: seem to have two possible reactions for an alkene with Br2. 1. Addition to the double bond yielding a dibromide. And/or 2. Substitution at allylic position.
Competing Reactions: Addition vs Substitution Substitution Addition Produced from Br2 at high temperature Br2 Br., not Br2 Alkene reacts directly with Br2 Happens at low Br atom concentrations. Low temperature keeps Br concentration low and thus favors addition. Alkene reacts preferentially with Br atoms if present. Favored by high Br atom concentrations. High temperature favors Br2 2Br and thus substitution.
Convenient allylic bromination For allylic substitution to occur we need both bromine atoms and Br2 Br: R-H + Br. R. + H-Br Br2: R. + Br2 R-Br + Br. Both Br and Br2 can be supplied from Br2 at high temperature or from NBS (N-bromosuccinimide). NBS
Allylic Rearrangement Expect bromination of but-2-ene to yield 1-bromo but-2-ene by replacing allylic hydrogen. But get rearranged product as well…. Major product, more stable with subsituted double bond.
Mechanism of Rearrangement Two different sites of reactivity More highly substituted alkene (more stable, recall hydrogenation data) is the major product
An interesting competition is occurring. Consider the allylic radical… The actual radical is a blending of these two structures. Secondary radicals are more stable than primary. This predicts most of the radical character at the secondary carbon, favoring this structure. But… But more highly substituted alkenes are more stable. This predicts most radical character at primary carbon favoring this structure. This appears to be the dominant factor leading to dominant product. But also….
Results of Calculation of Spin Densities in radical formed from 1-butene Blue is unpaired electron density. More at primary than secondary.
Anti Markovnikov addition of HBr Only with HBr, not HCl, HI
Mechanism of anti-Markovnikov Radical Addition of HBr Contrast radical and ionic addition of HBr Ionic Radical Not a Chain Process Chain Steps Common Concept: More stable intermediate formed, secondary radical or secondary carbocation
Sample Problem 4. A mixture of 1.6 g of methane and 1.5 g of ethane are chlorinated for a short time. The moles of methyl chloride produced is equal to the number of moles of ethyl chloride. What is the reactivity of the hydrogens in ethane relative to those in methane? Show your work. Solution: Recall: The amount of product is proportional to the number of hydrogens that can produce it multiplied by their reactivity. Number of hydrogens leading to methyl chloride = 1.6g * (1 mol/16 g) * (4 mol H/1 mol methane) = 0.40 mol H Number of hydrogens leading to ethyl chloride = 1.5 g * (1 mol/30 g) * (6 mol H/ 1 mol ethane) = 0.30 mol H 0.40 mol H * Rmethane = 0.30 mol H * Rethane Rethane/Rmethane = 0.4/0.3 = 1.3
How do we form the orbitals of the pi system… First count up how many p orbitals contribute to the pi system. We will get the same number of pi molecular orbitals. Three overlapping p orbitals. We will get three molecular orbitals.
If atomic orbitals overlap with each other they are bonding, nonbonding or antibonding Anti-bonding, destabilizing. Higher Energy But now a particular, simple case: distant atomic orbitals, on atoms not directly attached to each other. Their interaction is weak and does not affect the energy of the system. Non bonding If atoms are directly attached to each other the interactions is strongly bonding or antibonding. Bonding, stabilizing the system. Lower energy.
Molecular orbitals are combinations of atomic orbitals. They may be bonding, antibonding or nonbonding molecular orbitals depending on how the atomic orbitals in them interact. Example: Allylic radical Two antibonding interactions. Only one weak, antibonding (non-bonding) interaction. All bonding interactions.
Allylic Radical: Molecular Orbital vs Resonance Molecular Orbital. We have three pi electrons (two in the pi bond and the unpaired electron). Put them into the molecular orbitals. Note that the odd electron is located on the terminal carbons. Resonance Result Again the odd, unpaired electron is only on the terminal carbon atoms.
But how do we construct the molecular orbitals of the pi system? How do we know what the molecular orbitals look like? Key Ideas: For our linear pi systems different molecular orbitals are formed by introducing additional antibonding interactions. Lowest energy orbital has no antibonding, next higher has one, etc. 2 antibonding interactions 1 weak antibonding Interaction, “non-bonding” Antibonding interactions are symmetrically placed. 0 antibonding interactions This would be wrong.
Another example: hexa-1,3,5-triene Three pi bonds, six pi electrons. Each atom is sp2 hybridized. Have to form bonding and antibonding combinations of the atomic orbitals to get the pi molecular orbitals. Expect six molecular orbitals. # molecular orbitals = # atomic orbitals Start with all the orbitals bonding and create additional orbitals. The number of antibonding interactions increases as we generate a new higher energy molecular orbital.