Download

1 / 70
900 likes | 2.14k Views
10/2/16. AMYLOIDOSIS. Dr. Ksheera Cariappa Assistant Professor. CONTENT. Definition History Physical Nature Chemical Nature Fibril proteins Pathogenesis Classification Stains Organ morphology. Definition.

E N D
10/2/16 AMYLOIDOSIS Dr. Ksheera Cariappa Assistant Professor
CONTENT • Definition • History • Physical Nature • Chemical Nature • Fibril proteins • Pathogenesis • Classification • Stains • Organ morphology
Definition Amyloidosis is a term used for a group of diseases characterized by extracellular deposition of fibrillary proteinaceous substance called amyloid having characteristic morphological appearance, staining properties and physical structure, but with variable protein or biochemical composition.
HISTORY • 1854 Rudolph Virchow named it amyloid based on color after staining these proteins with iodine and sulfuric acid. • Meaning cellulose or starch • Amyloid = Starch like
Physical Nature Amyloid fibril protein occurs in tissue deposits as insoluble, rigid, non-branching fibrils 7-to 10 nm in dm When analysed by X-ray diffraction, the fibrils exhibit a characteristic cross Beta diffraction pattern
Chemical Nature • Pentagonal molecule • 95% Protein Fibril • 5% Glycoprotein P component
Fibril proteins • Fibrils are delicate, randomly dispersed, non branching, each measuring 7.5 – 10 nm in diameter & indefinite length. • Each fibril- double helix • X-ray crystallography and infra red spectroscopy- cross B pleated sheet configuration( staining properties of amyloid with congo red) • Chemical analysis- heterogenous • Major forms AL( Amyloid light chain) protein AA( Amyloid associated) protein Others
Classification • Based on cause - Primary and Secondary • Based on extent of amyloid deposition -Systemic & Localized Systemic (Generalized) Primary ( AL) Secondary(AA) Haemodialysis associated(AB2M) Heredofamilial (ATTR,AA,other) Localized amyloidosis Senile cardiac(ATTR) Senile cerebral(Ab) Endocrine( hormone precursors) Tumor forming(AL)
Primary amyloidosis • AL ( light chain proteins) • Associated diseases- Plasma cell dyscrasiase.g multiple myeloma, B cell lymphoma, others • Organ distribution- kidney, heart, bowel, nerves • Stains to distinguish- congophiliaperisists after permanganate treatment of section; specific immunostains anti L, anti K. • Pathogenesis- Stimulus----- Monoclonal B cell proliferation----- Excess Igs & light chains-----partial degradation------Insoluble AL fibril
Idiopathic Multiple myeloma B cell lymphoma Other plasma cell dyscrasias Stimulus Monoclonal B cell proliferation Soluble precursor protein Excess Igs( Intact Igs, L and K) Macrophage Partial degradation Non fibrillar components AL Amyloid Insoluble fibril protein
(i) PRIMARY AMYLOIDOSIS (AL TYPE) (IMMUNOCYTEDYSCRASIAS) Most commonform Associated with plasma celldyscrasias Monoclonal plasma cells synthesize either λ or κ chains, that gives a M spike on electrophoresis Due to small molecular size – Bencejone’s protein is found inurine
Secondary amyloidosis • AA (amyloid associated protein) derived from larger precursor protein SAA. (Serum Amyloid Associated) • Associated disease chronic inflammation e.g infections( TB, leprosy, osteomyelitis, bronchiectasis), autoimmune diseases ( rheumatoid arthritis, IBD), cancers ( RCC, Hodgkin`s disease), FMF • Organ distribution-kidney, liver, spleen, adrenals • Stains to distinguish- congophilia disappears after permanganate treatment of section, specific immunostains anti AA. • Pathogenesis- Stimulus----- chronic inflammation-----activation of macrophages------cytokines(IL 1,6)----partial degradation----AEF (Amyloid Enhancing Factor)-----Insoluble fibril
Chronic infections Ch non infectious inflammation Cancer Familial Mediterranean fever Stimulus Activation of macrophage Soluble precursor protein SAA protein by liver cells In reticulo-endothelial cell Partial degradation Non fibrillar components AA Amyloid Insoluble fibril protein
SECONDARY AMYLOIDOSIS (AATYPE) • Secondary to infective conditions like TB, bronchiectasis, chronicosteomyelitis • Inflammatory conditions like Rheumatoid arthritis, Ankylosing spondylitis, inflammatory bowel disease, Crohn’s and ulcerativecolitis • skin popping in heroinabusers • non immunocyte derived tumors like renal cell carcinomas andHodgkin’s
(iii) HEMODIALYSIS ASSOCIATED AMYLOIDOSIS (aβ 2 MICROGLOBULIN) In patients with renal disease, β 2 microglobulin is present in highconcentrations Also this protein is not filtered through the dialysismembranes It gets deposited in synovium / tendons /joints May lead to carpal tunnelsyndrome
A cell block prepared from the knee synovial fluid of a patient with dialysis-related beta-2 microglobulin amyloidosis showed amorphous material that stained with Congo red.
(iv) FAMILIAL MEDITERRANEAN FEVER (AAPROTEIN) FAMILIAL MEDITERRANEAN FEVER is a hereditary genetically restricted disease commonly found among Jews originating from North African countries, Armenians, Turks and Arabs. FMF is recognized by two independentmanifestations: 1) acute, short-lived painful, bouts of stomach pain (peritonitis) or pleuritiswith fever 2)nephropathic amyloidosis,which canleadtoterminalrenalfailure evenata young age. The cause is a lack of pyrin, a neutrophil protein which slows down neutrophils when enough have reached anarea Lacking pyrin, neutrophils mob body cavities every once in awhile. Colchicine, famous for its ability to slow down neutrophils (as in acute gout), controls the attacks and prevents the dreaded complication of secondaryamyloidosis
(v) FAMILIAL AMYLOIDOTIC POLYNEUROPATHY (mutantaTTR) Familial amyloid neuropathies(FAP) are a group of familial systemic amyloidoses with involvement of peripheral nerves. The most common FAP is caused by an autosomal dominant mutation of the transthyretin gene on18q11. The mutant protein is deposited in the form of amyloid and damages peripheral nerves, the heart, kidneys, gastrointestinal tract, and otherorgans. In nerves, amyloid damages first and most severely small fibers, causing loss of pain and temperature sensation and autonomicdysfunction. Transthyretin is produced in the liver. Liver transplantation arrests the progression ofthe disease.
(vi) SENILE SYSTEMIC AMYLOIDOSIS/SENILE CARDIAC AMYLOIDOSIS) (Normal aTTR) Elderlypatients Present with restrictive cardiomyopathy andarrythmias CARDIAC (HEART) AMYLOIDOSIS] is usually seen in two clinical settings, eitheraspart of systemic amyloidosis or isolated senile cardiac amyloidosis not involving any other organs. The systemic amyloidosis is generally seen in patients with underlying plasma cell dyscrasia. The senile cardiac amyloidosis is due to deposition of normal form of transthyretin. Amyloid appears as light-pink hyaline extracellular deposits (left arrow) displacing cardiac myocytes (rightarrow).
(vii) DEPOSITION OF AMYLOID IN ENDOCRINE DISORDERS / TUMORS PRODUCINGHORMONES a. MEDULLARY CARCINOMA OF THYROID (a CAL)(CALCITONIN) Amyloid deposition in medullary cathyroid MTC can be remembered by the 3 Ms: aMyloid. Median node dissection. MEN 2A & MEN2B
b. IN PATIENTS WITH DIABETES MELLITUS (a IAPP) (islet associatedpolypeptide) The amyloid is deposited in the Islets of Langerhans, the endocrinepancreas
ISOLATED ATRIAL AMYLOIDOSIS (a ANF) (atrial natriureticfactor) • Deposition of atrial natriureticfactor • SENILE CEREBRAL AMYLOIDOSIS (a βprotein) • Cerebral amyloid angiopathy (CAA) is a fundamental part of the pathology of many disorders causing dementia and/or cerebral haemorrhage. • In Alzheimer's disease (AD), CAA is due to the deposition of amyloid alpha protein (Abeta) within the adventitia and media of leptomeningeal and brain parenchymalarteries. • In vessels affected by CAA, local muscle and elastic elements are lost and replacedby • amyloid fibrils, thereby weakening the overall structure of the vessel. Consequently, CAA predisposes towards cerebral infarction and cerebralhaemorrhage,
STAINING CHARACTERSTICS OF AMYLOID • 1. Stain on gross:- Oldest method since the time of Virchow for demonstrating amyloid on cut surface of a gross specimen, or on the frozen/ paraffin section is iodine stain. • Lugol’s iodine imparts mahogany brown colour to the amyloid- containing area which on addition of dilute sulfuric acid turns blue. • This starch- like property of amyloid is due to AP component, a glycoprotein, present in all forms of amyloid.
2. H & E • Appears as extracellular, homogeneous, structure less and eosinophilic hyaline material, especially in relation of blood vessels. • However, if the deposits are small, they are difficult to detect by routine H and E stains. Besides, a few other hyaline deposits may also take up pink colour.
3. METACHROMATIC STAINS (ROSANILINE DYES) • Amyloid has the property of metachromasia i.e. the dye reacts with amyloid and undergoes a colour change. • Metachromatic stains employed are rosaniline dyes such as methyl violet and crystal violet which impart rose- pink colour to amyloid deposits.
However, small amounts of amyloid are missed, mucin also has metachromasia property and hence aqueous mountants are required for seeing this preparation. • Therefore, this method has low sensitivity and lacks specificity.
METACHROMATIC STAINS Metachromatic rose-pink staining amyloid inglomerulus
4. CONGO RED AND POLARISED LIGHT • All types of amyloid have affinity for Congo red stain; therefore this method is used for confirmation of amyloid of all type. • Stain may be used on gross specimens and microscopic sections; amyloid of all types stains pink red colour. • If the stained section is viewed in polarised light, the amyloid characteristically shows apple- green birefringence due to cross- beta- pleated sheet configuration of amyloid fibrils.
The stain can also be used to distinguish between AL and AA amyloid.. • After prior treatment with permanganate or trypsin on the section, Congo red stain is repeated- in the case of primary amyloid (AL amyloid), the Congo red positivity (congophilia) persists, while it turns negative for Congo red in secondary amyloid (AA amyloid).
Congo red dye can also be used as an in vivo test. • A known quantity of Congo red dye may be injected intravenously in living patients. • If amyloidosis is present, the dye gets bound to amyloid deposits and its levels in blood rapidly declines. • The test is, however, not popular due to risk of anaphylaxis to the injected dye.
CONGO RED AND POLARISED LIGHT • CONGO RED STAIN • CONGO RED STAIN IN POLARISED LIGHT
5. FLUORESCENT STAINS • Fluorescent stain Thioflavin - T binds to amyloid and fluoresce yellow under ultraviolet light i.e. amyloid emits secondary fluorescence. • Thioflavin- S is less specific.
FLUORESCENT STAIN: Amyloid deposited in wall of cerebral blood vessel in alzheimer’s– ThioflavinTstain
6. IMMUNOHISTOCHEMISTRY • More recently, type of amyloid can be classified by immunohistochemical stains. • Various antibodies stain against the specific antigenic protein type of amyloid and are commercially available. • However, most useful in confirmation for presence of amyloid of any type is anti- AP stain; others for determining the biochemical type of amyloid include anti- AA, anti- lambda, anti- kappa antibody stains etc.
7. NON- SPECIFIC STAINS • A few other stains have been described for amyloid at different times but they lack specificity. • These are as under:- • i) Periodic acid Schiff (PAS):- This is used for demonstration of carbohydrate contents of amyloid but shows variable positivity and not specific.
ii) Standard toluidine blue:- This method gives orthochromatic blue colour to amyloid which under polarising microscopy produces dark red birefringence. • However, there are false positive as well as false negative results; hence not recommended.
TOLUIDINE BLUE STAIN Perimyocyticamyloid deposition in semi-thin plastic-embedded myocardium. This micrograph illustrates perimyocytic amyloiddeposits (pale purple-blue) surrounding and distorting individual myocytes (dark purple-blue). The toluidine blue sections are used to map the areas to be examined in ultrathin sections in electronmicroscopy.
iii) Alcian blue:- It imparts blue- green colour to amyloid positive areas and for mucopolysaccharide content in amyloid but uptake of dye is poor and variable. Microscopy of cardiac tissue from autopsy demonstrates amyloid deposition between cardiac myocytes as homogeneous light pink material (left). SulfatedAlcianblue staining shows extensive amyloid deposition as green amorphous material(right)
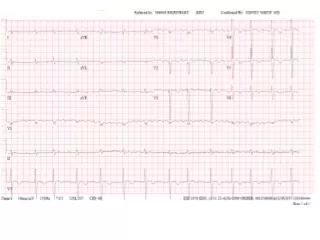
AMYLOIDOSIS DIAGNOSIS • Need to demonstrate amyloid in the tissues. • Rectal biopsy, kidney biopsy, gingival biopsy, fat pad • FNAC. • Serum and urine protein electrophoresis and immunoelectrophoresis. • Bone marrow biopsy. • EKG. Cardiac ultrasound.
Sites of amyloid deposition Contacts between the vascular spaces and parenchymal cells, in the extracellular matrix and within the basement membranes of blood vessels. Gross Affected organ usually enlarged, pale and rubbery. C/S firm, waxy and translucent, positive staining with the iodine test. Microscopy Deposits seen in extracellular location, walls of blood vessels
1. Amyloidosis of kidney Amyloidosis of kidney– most cases of secondary amyloidosis and in one third cases of primary amyloidosis. GROSS: Kidneys normal sized, enlarged or terminally contracted. Cut surface- pale,waxy and translucent. MICROSCOPY: Amyloid deposition occurs primarily in the glomeruli, later affects tubules and walls of small arterioles and venules.