Download

1 / 26
270 likes | 455 Views
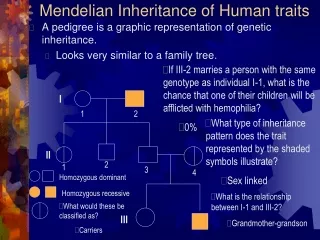
Human Normal Mendelian Traits. a.k.a. (classic) Genetic Systems in Humans. Blood groups (system).

E N D
Human Normal Mendelian Traits a.k.a. (classic) Genetic Systems in Humans
Blood groups (system) A blood type (also called a blood group) is a classification of blood based on the presence or absence of inherited antigenic substances on the surface of red blood cells (RBCs). These antigens may be proteins, carbohydrates, glycoproteins, or glycolipids, depending on the blood group system. Some of these antigens are also present on the surface of other types of cells of various tissues. Several of these red blood cell surface antigens can stem from one allele (or very closely linked genes, e.g. A1, A2, B1) and collectively form a blood group system.Blood types are inherited and represent contributions from both parents. A total of 32 human blood group systems are now recognized by the International Society of Blood Transfusion (ISBT), although only the ABO and Rh systems are clinically significant. Two or more alleles are fully dominant (Iᴬ, Iᴮ) When present together they are both expressed ABO Blood Groups exhibit co-dominance (AB), AD (A,B heterozygous) & recessiveness (O)
Individuals with group A blood have red blood cells with antigen A on their surface. Produce antibodies against antigen B, antibody B. Therefore, a group A person can only receive blood from people in groups A or O Individuals with group O blood have red blood cells with neither antigen A or B. Produce antibodies A and B against both types of antigens. A group O person can only receive blood from group O (universal donor). Individuals with group B blood have red blood cells with antigen B on their surface. Antibodies A are produced in their serum. A group B person can only receive blood from people in groups B or O, preferably B. Individuals with group AB blood have red blood cells with both antigens A and B. Do not produce antibodies A or B against either antigen in their serum. A person with type AB blood can receive blood from any group (preferably AB) but cannot donate blood except to another AB (universal recipient).
Bombay Phenotype • This is an extremely rare ABO group which derives the name "Bombay" because it was first discovered to exist among some people living in the region of Bombay, India. Although the group is more likely to occur in East Indians, it is a very rare group even among this population. Also, it is not restricted to East Indians and has been found to exist in Caucasians, Negroes, Japanese, etc. • Inheritance:The Bombay group (Oh) results from the inheritance of two rare recessive h genes which occur at a locus other than the ABO gene locus. • Because the h gene is very rare, Bombays often result from consanguineous matings in which parents are blood relatives (e.g., first cousins). Whenever inbreeding occurs, the proportion of rare homozygotes increases in frequency. • Their red cells lack ABH antigens and their sera contain anti-A and anti-B and anti-H. • The anti-H would not be detected in the ABO group but would be detectable in pretransfusion tests, e.g., their sera would agglutinate group O screen cells and group O donor cells, which have the H antigen. • In blood transfusion:Bombay people would be incompatible when crossmatched with red cells of all normal ABO groups (groups O, A, B and AB). • If they require blood transfusion, they must receive blood from another Bombay. Donors must be sought among their blood relatives (especially siblings) or from the rare donor file maintained by the Red Cross. Person #1 and #2 are consanguineous. Their child is a Bombay and has inherited an A gene and a B gene from her parents and has transmitted them to her offspring (#7 and #8). Therefore, Bombay people can have A and B genes, even though they cannot make ABH antigens because they lack the H gene.
The biosynthesis of the H antigen and the A and B antigens involves a series of enzymes (glycosiltransferases) that transfer monosaccharides. The resulting antigens are oligosaccharide chains, which are attached to lipids and proteins that are anchored in the RBC membrane. The function of the H antigen, apart from being an intermediate substrate in the synthesis of ABO blood group antigens, is not known although it may be involved in cell adhesion. Fortunately people who lack of the H antigen do not suffer from any deleterious effects, and being H-deficient is only an issue if they were to need a blood transfusion because they would require H-deficient blood.
An ABO/ABH particularity - Secretor/Non-Secretor system • Individuals who present the erythocytes ABO antigens in other tissues (saliva, plasma, perspiration) or tumors are called secretory and represent the vast majority (75%) of the population. • Individuals who present the ABO antigens only on the erythocytes are called non-secretory and represent 25% of the population. • Genes responsible of this character are Se (dominant) and se (recessive)
Non Secretors Variation what it means • Have lower level of intestinal phosphatase for protein digestion • Like blood type A and AB to have higher levels of Factor and Factor VIII • Non secretors develop Cardiovascular Disease from metabolic syndrome insulin resistance and type ii diabetes. • Non Secretors have higher levels of CRP (inflammatory markers) than secretors. • Foreign bacteria invade the system more readily • Have a higher risk of cancer than secretors • Are more susceptible to Candida • Immune system is geared differently due to the presence of ABO antigens inside the cell • Are susceptible to dental caries • Are sensitivity to bacteria which cause ulcers • Have a relatively high risk for the development of inflammatory bowel problems • Are at a higher risk of alcohol related disorders. • Have different dietary requirements and supplemental regime
Rh blood group system • The Rh system is the second most significant blood-group system in human-blood transfusion with currently 50 antigens. The most significant Rh antigen is the D antigen, because it is the most likely to provoke an immune system response of the 5 main Rh antigens (C,D,E,F,G) • The presence or absence of the Rh antigens is signified by the + or − sign, so that for example the A− group does not have any of the Rh antigens. • Rh− blood types are much less frequent in Asian populations (0.3%) than they are in White (15%). • Rh disease (also known as Rhesus isoimmunisation, Rh (D) disease, Rhesus incompatibility, is one of the causes of hemolytic disease of the newborn (HDN). The disease ranges from mild to severe, and typically occurs only in some second or subsequent pregnancies of Rh negative women where the fetus's father is Rh positive, leading to a Rh+ pregnancy. During birth, the mother may be exposed to the infant's blood, and this causes the development of antibodies, which may affect the health of subsequent Rh+ pregnancies. In mild cases, the fetus may have mild anaemia with reticulocytosis. In moderate or severe cases the fetus may have a more marked anaemia and erythroblastosis (erythroblastosisfetalis). When the disease is very severe it may cause haemolytic disease of the newborn (HDN), hydropsfetalis or stillbirth. • Rh disease is generally preventable by treating the mother during pregnancy or soon after delivery with an intramuscular injection of anti-RhD immunoglobulin (Rho(D) immune globulin). The RhD protein is coded for by the RHD gene.
MNS blood antigen system • The MNS antigen system is a human blood group system based upon two genes (glycophorin A and glycophorin B) on chromosome 4. • There are currently 46 antigens in the system,but the five most important are called M, N, S, s, and U. • The system can be thought of as two separate groups: • the M and N antigens are at one location and • S, s, and U are on a closely related location. • The two groups are very closely located together on chromosome 4 and are inherited as a haplotype. • M+ and N+ RBCs are common (75% of population) and M+N+ cells are the most common genotype (50% of population). These antigens were an early discovery and are some of the oldest blood antigens known after the ABO system. • Anti-M and anti-N antibodies are rarely associated with transfusion reactions. • The S antigen is relatively common (~55% of the population) and the s antigen is very common (~89% of the population). Anti-S and anti-s can cause rarely hemolytic transfusion reactions and hemolytic disease of the newborn. • The U antigen is a high incidence antigen, occurring in more than 99.9% of the population. The U was originally short for "Universal", though this is not the case. U negative RBCs can be found in people of African descent. This mutation in red cell surface structure also makes the RBCs S- and s-. Anti-U has been occasionally associated with both hemolytic transfusion reactions and hemolytic disease of the newborn.
Hemoglobin genetic system • Hemoglobin in the blood carries oxygen from the lungs to the rest of the body (tissues) where it releases the oxygen to burn nutrients to provide energy to power the functions of the organism, and collects the resultant carbon dioxide to bring it back to the respiratory organs to be dispensed from the organism. • In mammals, the protein makes up about 97% of the red blood cells' dry content, and around 35% of the total content (including water). • In adult humans, the most common hemoglobin type is a tetramer (which contains 4 subunit proteins) called hemoglobin A, consisting of two α and two β subunits non-covalently bound, each made of 141 and 146 amino acid residues, respectively. This is denoted as α2β2 • In human infants, the hemoglobin molecule is made up of 2 α chains and 2 γ chains. The gamma chains are gradually replaced by β chains as the infant grows. This is fetal hemoglobin denoted α2 γ2 Each subunit is composed of a protein chain tightly associated with a non-protein heme group. A heme group consists of an iron (Fe) ion (charged atom) held in a heterocyclic ring, known as a porphyrin.
Adult hemoglobin Hemoglobin A (left) is the most intensively studied of the hemoglobin molecules.
Pathologic hemoglobins • In sickle cell hemoglobin (HbS) glutamic acid in position 6 (in beta chain) is mutated to valine. • This change allows the deoxygenated form of the hemoglobin to stick to itself.
HbS - Sickle cell anemia (2) • Sickle-cell disease may lead to various acute and chronic complications, several of which have a high mortality rate • Sickle cell crisis The term "sickle cell crisis" is used to describe several independent acute conditions occurring in patients with sickle cell disease. Sickle cell disease results in anemia and crises that could be of many types including the vaso-occlusive crisis, aplastic crisis, sequestration crisis, haemolytic crisis and others. Most episodes of sickle cell crises last between five and seven days.Although infection, dehydration, and acidosis (all of which favor sickling) can act as triggers, in most instances no predisposing cause is identified. • Vaso-occlusive crisis The vaso-occlusive crisis is caused by sickle-shaped red blood cells that obstruct capillaries and restrict blood flow to an organ, resulting in ischaemia, pain, necrosis and often organ damage. The frequency, severity, and duration of these crises vary considerably. Painful crises are treated with hydration, analgesics, and blood transfusion; pain management requires opioid administration at regular intervals until the crisis has settled. For milder crises, a subgroup of patients manage on NSAIDs (such as diclofenac or naproxen). For more severe crises, most patients require inpatient management for intravenous opioids; patient-controlled analgesia (PCA) devices are commonly used in this setting. Vaso-occlusive crisis involving organs such as the penis or lungs are considered an emergency and treated with red-blood cell transfusions. Diphenhydramine is sometimes effective for the itching associated with the opioid use. Incentive spirometry, a technique to encourage deep breathing to minimise the development of atelectasis, is recommended.
HbS - Sickle cell anemia (3) • Splenic sequestration crisis Because of its narrow vessels and function in clearing defective red blood cells, the spleen is frequently affected. It is usually infarcted before the end of childhood in individuals suffering from sickle-cell anemia. This autosplenectomy increases the risk of infection from encapsulated organisms;preventive antibiotics and vaccinations are recommended for those with such asplenia. Splenic sequestration crises: are acute, painful enlargements of the spleen, caused by intrasplenic trapping of red cells and resulting in a precipitous fall in hemoglobin levels with the potential for hypovolemic shock. Sequestration crises are considered an emergency. If not treated, patients may die within 1–2 hours due to circulatory failure. Management is supportive, sometimes with blood transfusion. These crises are transient, they continue for 3–4 hours and may last for one day. • Aplastic crisis Aplastic crises are acute worsenings of the patient's baseline anaemia, producing pallor, tachycardia, and fatigue. This crisis is normally triggered by parvovirus B19, which directly affects erythropoiesis (production of red blood cells) by invading the red cell precursors and multiplying in them and destroying them.Parvovirus infection nearly completely prevents red blood cell production for two to three days. In normal individuals, this is of little consequence, but the shortened red cell life of sickle-cell patients results in an abrupt, life-threatening situation. Reticulocyte counts drop dramatically during the disease (causing reticulocytopenia), and the rapid turnover of red cells leads to the drop in haemoglobin. This crisis takes 4 days to one week to disappear. Most patients can be managed supportively; some need blood transfusion. • Haemolytic crisis Haemolytic crises are acute accelerated drops in haemoglobin level. The red blood cells break down at a faster rate. This is particularly common in patients with co-existent G6PD deficiency. Management is supportive, sometimes with blood transfusions.
Other types of pathologic hemoglobin • Hemoglobin C (α2βC2) - Another variant due to a variation in the β-chain gene; glutamic acid in position 6 (in beta chain) is mutated to lysine. This variant causes a mild chronic hemolytic anemia. • Hemoglobin E (α2βE2) - Another variant due to a variation in the β-chain gene; glutamic acid in position 26 (in beta chain) is mutated to lysine. This variant causes a mild chronic hemolytic anemia.
Abnormal quantitative synthesis of hemoglobin • Thalassemias are genetic disorders caused by a reduction of synthesis of globin chains. • α-Thalassemias The α thalassemias involve the genes HBA1 and HBA2,inherited in a Mendelian recessive fashion. There are two gene loci and so four alleles. α- Thalassemias result in decreased alpha-globin production, therefore fewer alpha-globin chains are produced, resulting in an excess of β chains in adults and excess γ chains in newborns. The excess β chains form unstable tetramers (called Hemoglobin H or HbH of 4 beta chains), which have abnormal oxygen dissociation curves. • β-Thalassemias Beta thalassemias are due to mutations in the HBB gene on chromosome 11,also inherited in an autosomal-recessive fashion. The severity of the disease depends on the nature of the mutation. Mutations are characterized as either βo or β thalassemia major if they prevent any formation of β chains, the most severe form of β thalassemia. Also, they are characterized as β+ or β thalassemia intermedia if they allow some β chain formation to occur. In either case, there is a relative excess of α chains, but these do not form tetramers: Rather, they bind to the red blood cell membranes, producing membrane damage, and at high concentrations they form toxic aggregates. • Delta (δ) thalassemia As well as alpha and beta chains present in hemoglobin, about 3% of adult hemoglobin is made of alpha and delta chains. Just as with beta thalassemia, mutations that affect the ability of this gene to produce delta chains can occur. Thalassemias have an autosomal recessive pattern of inheritance
The genetic system of Transferrins • Transferrins are iron-binding blood plasma glycoproteins that control the level of free iron in biological fluids. They are β1-globulins playing a part in binding the plasma iron and transporting it to all the tissues. • Transferrin glycoproteins bind iron very tightly, but reversibly. • Human transferrin is encoded by the TF gene, which has 3 alleles (TfC, TfB and TfD) • The TF alleles exhibit co-dominance and can generate 6 genotypes:
The genetic system of Haptoglobin • Haptoglobin (abbreviated as Hp) is the protein that in humans is encoded by the HP gene. In blood plasma, haptoglobin binds free hemoglobin (Hb) released from erythrocytes with high affinity and thereby inhibits its oxidative activity. The haptoglobin-hemoglobin complex will then be removed by the reticuloendothelial system (mostly the spleen). In clinical settings, the haptoglobulin assay is used to screen for and monitor intravascular hemolytic anemia. • Haptoglobin is produced mostly by hepatocytes but also by other tissues: e.g., skin, lung, and kidney. In addition, the haptoglobin gene is expressed in human adipose tissue. • Haptoglobin, in its simplest form, consists of two α- and two β-chains, connected by disulfide bridges. The chains originate from a common precursor protein, which is proteolytically cleaved during protein synthesis. • Hp exists in two allelic forms in the human population, so-called Hp1 and Hp2, the latter one having arisen due to the partial duplication of Hp1 gene (evolutionary). • Three genotypes of Hp, therefore, are found in humans: Hp1-1, Hp2-1, and Hp2-2. • Hp of different genotypes have been shown to bind hemoglobin with different affinities, with Hp2-2 being the weakest binder.
Acid phosphatase genetic system • Acid phosphatase (PA) is an enzyme present especially in the prostate (isoenzyme PAP)but also in serum, erythrocytes, leucocytes, thrombocytes, liver, spleen, kidney, endocrine glands. • PA plays an important part in diagnosing the metastasis of prostate cancer, but also in bone metastasis of colon cancer and several other cancers: breast, adrenal gland and pulmonary. Also in thrombosis and pulmonary embolies. • The gene responsible for the synthesis of PA protein is called P which has 3 alleles: a,b,c
The Gustatory system • The gustatory system is the sensory system for the sense of taste. • The gustatory system allows humans to distinguish between safe and harmful food. Bitter and sour foods we find unpleasant, while salty, sweet, and meaty tasting foods generally provide a pleasurable sensation. The five specific tastes received by gustatory receptors are salty, sweet, bitter, sour, and umami, which means “savory” or “meaty” in Japanese. • The gustatory status is defined by the ability of a person to taste the bitterness of PTC (phenylthiocarbamide). • The gene responsible is called G and has two alleles (G-dominant and g-recessive) and therefore there are 3 possible genotypes: • The most frequent in Europe is G/G • Clinically, Gustatory phenotypes are at larger risk of developing Basedow-Graves disease (thyrotoxic goiter); meanwhile Non-gustatory phenotypes appear more at risk for adenomatous goiter