Download
1 / 14
140 likes | 324 Views
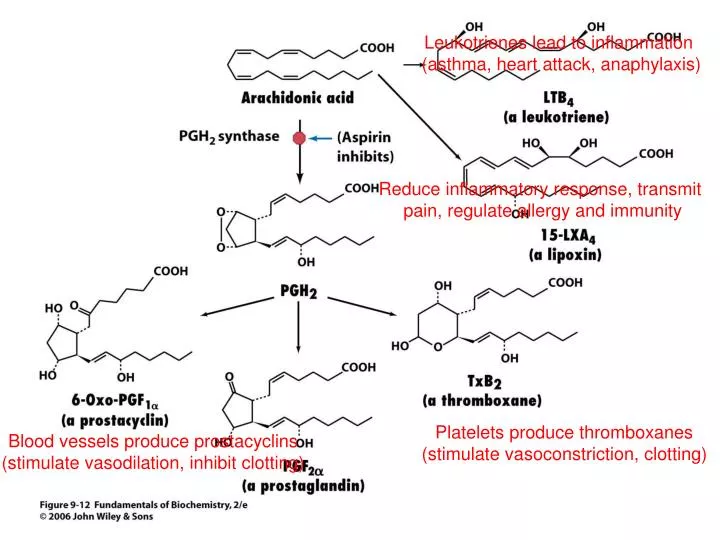
Leukotrienes lead to inflammation (asthma, heart attack, anaphylaxis). Reduce inflammatory response, transmit pain, regulate allergy and immunity. Platelets produce thromboxanes (stimulate vasoconstriction, clotting). Blood vessels produce prostacyclins

E N D
Leukotrienes lead to inflammation (asthma, heart attack, anaphylaxis) Reduce inflammatory response, transmit pain, regulate allergy and immunity Platelets produce thromboxanes (stimulate vasoconstriction, clotting) Blood vessels produce prostacyclins (stimulate vasodilation, inhibit clotting)
Arachidonic acid • Eicosanoid (C20) precursor from dietary essential polyunsaturated fatty acids (linoleic acid) • Stored as C2-ester of phospholipids (released by phospholipase A2) • Hormone-like molecules, decompose within seconds or minutes (have very local effects) • Pain, fever, coagulation, blood pressure, and reproduction
Prostaglandins • Prostaglandin H2 synthase forms a cyclopentane ring in linear arachidonic acid • Enzyme has 2 catalytic activities: • Cyclooxygenase (adds two O2) • Peroxidase (converts OOH group to OH) • Commonly called COX • Aspirin is an irreversible inhibitor of COX (inactivates enzyme by acetylating active site Ser and blocking active site from reacting with substrate • Analgesic, antipyretic, anti-inflammatory Ser-ÖH
COX • NSAIDs (NonSteroidal Anti-Inflammatory Drugs) • Acetaminophen and ibuprofen are noncovalent inhibitors of COX • COX-1 and COX-2 isoforms (60% identical) • COX-2 inhibitors lack side effects of other NSAIDS • COX-1 expressed ubiquitously, maintains homeostasis • COX-2 expressed in certain tissues during inflammation response and is responsible for elevated prostaglandin levels • Aspirin and ibuprofen are nonspecific NSAIDs, have side effects (gastrointestinal ulceration)
COX-2 • Structure-based drug design of selective COX-2 inhibitors • COX-2 active site ~20% larger than COX-1 (make a bigger inhibitor that cannot fit into COX-1 site) • Rofecoxib (Vioxx) popular and effective, but withdrawn due to unanticipated cardiac side effects (mechanism may involve inhibition of prostacyclin synthesis (leaving thromboxane synthesis less affected) • Acetaminophen effective, but low affinity for COX-1,2 (binds COX-3, which is expressed in the central nervous system) COX inhibitor
Drug Design • Drug Discovery: How? Screening large numbers of compounds for inhibition of enzymatic activity or receptor signaling Measure KI(or KI’) Good lead compound has KI < 1mM Why is high affinity necessary? -specificity!!!!! -dose!!!!!!! Design related compounds using combinatorial chemical techniques
Structure-based design • X-ray, NMR • What does the active site look like empty and with the substrate in it? • Look at structural and electrostatic properties of active site and try to better fit/fill it. For a candidate… • Quantum mechanical calculation of charge distribution • Docking simulations • Determine structure of complex, revise inhibitor structure and re-assay KI
Bioavailability & Toxicity To cause desired response: • Drug must arrive at high enough concentration • Drug must arrive to the location of the target protein Oral drugs (cheapest) • Acid-stable (stomach) • Membrane-permeable (gut-blood transfer, so can’t be highly charged)) • Don’t bind tightly to other things (lipophilic drugs sequestered in membrane/adipocytes) • Survive detoxifying enzymes in the liver (portal vein drains intestine directly to liver) • Avoid rapid excretion by kidneys • Must pass from capillaries to tissues • (for brain) must pass blood-brain barrier, which blocks polar substances • (for intracellular protein) must pass plasma (and other) membrane(s) Protein drugs poor oral drugs (acid, pepsin, trypsin, immune system, etc.)
Bioavailability & Toxicity How drug interacts with barriers is pharmacokinetics (Absorption, Distribution, Metabolism, Excretion measurements) Bioavailability (extent to which it reaches proper site) depends on dose and pharmacokinetics Best drugs a compromise: not too polar or lipophilic, neutral at pH 6-8 (pass through membrane in uncharged state) Drugs with low KI for target are likely to be more specific and have fewer side effects
Clinical trials After in vitro (test tube) and in vivo (animal) studies: Phase I: test safety, dosage range and method (20-100 healthy volunteers, or if toxic drug then test very sick people) Phase II: efficacy against target disease (100-500 volunteer patients). Refine dosage, check for side effects. Single-blind tests (docs know, patients don’t) Control substance is a placebo (ethical caveat) Phase III: Monitor adverse effects from long-term use, confirm efficacy (1000-5000 patients) through statistical analysis of double-blind, placebo-controlled tests (double blind removes bias from subjective judgments of investigators…you see what you want to see…)
Clinical trials Few drug candidates survive preclinical testing (~5/5000, ~3 years) Clinical trials 7-10 years, most fail in Phase II ~$500 million to bring drug to market!!! Most difficult issue is identifying rare side effects (1/10000) and long-term effects Out of Control, by Celia Farber (HIV clinical trial article… holiday reading) http://www.harpers.org/archive/2006/03/0080961
Cytochromes P450 Well-tolerated drugs can be dangerous for others… Genetic differences among individuals Different disease state Other drugs Sex Age Environmental factors
Cytochromes P450 Detoxify xenobiotics (embedded in ER membrane) Superfamily of heme-containing enzymes in nearly all living organisms Fe(II), CO-bound enzyme absorbs at 450nm Humans have ~100 isozymes (isoforms) Monooxygenases (Fe undergoes reversible redox-state change during catalytic cycle) RH + O2 + 2H+ + 2e- ROH + H2O e- from NADPH to the P450 heme via cytochrome P450 reductase Oxidize lipophilic compounds for conjugation to glucuronic acid or sulfate.