Download
1 / 55
590 likes | 779 Views
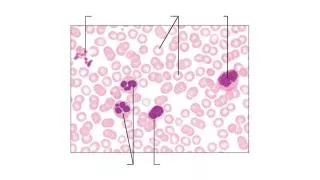
Platelets. Platelets are fragments of cytoplasm of bone marrow megakaryocytes Count 150 – 400 x 10 9 /l Major role in coagulation. Non-Malignant Platelets Disorders. Hemostasis Primary vs. Secondary vs. Tertiary. Primary Hemostasis Platelet Plug formation

E N D
Platelets • Platelets are fragments of cytoplasm of bone marrow megakaryocytes • Count 150 – 400 x 109/l • Major role in coagulation
HemostasisPrimary vs. Secondary vs. Tertiary • Primary Hemostasis • Platelet Plug formation • Dependent on normal platelet number & function • Initial manifestation of clot formation • Secondary Hemostasis • Activation of the clotting cascade • Deposition & Stabilization of fibrin • Tertiary Hemostasis • Dissolution of the fibrin clot • Dependent on Plasminogen Activation
PLT disorders: • Quantitative disorders: • “Penias” & “Philias” & Malignancies. • Qualitative disorders - function disorders • Congenital • Thrombasthenia, Bernad Soulier syndrome. • Acquired • Aspirin, Heparin. • Uremia, diabetes, hyperglobulinemia.
Quantitative disorders Abnormal distribution Dilution effect Decreased production Increased destruction Qualitative disorders Inherited disorders (rare) Acquired disorders Medications Chronic renal failure Cardiopulmonary bypass Classification of platelet disorders
Quantitative PLT disorders: • Decreased Production: • Marrow failures - aplasia, malignancy, drugs • Increased consumption: • DIC, TTP, Septicemia • Increased destruction: • Autoimmune, drugs, SLE, viral infections. • Dilutional thrombocytopenia: • Massive transfusions of stored blood.
Thrombocytopenia Immune-mediated Idioapthic Drug-induced Collagen vascular disease Lymphoproliferative disease Sarcoidosis Non-immune mediated DIC Microangiopathic hemolytic anemia
Associated with bleeding Immune-mediated thrombocytopenia (ITP) Most others Associated with thrombosis Thrombotic thrombocytopenic purpura Heparin-associated thrombocytopenia Trousseau’s syndrome DIC Classification of thrombocytopenia
ITP • idiopathic autoimmune platelet destruction • #1 cause of isolated thrombocytopenia in otherwise healthy young persons • a diagnosis of exclusion
ITP: Clinical features • occurs in any age or sex, but typically young female • can be preceeded by viral infection • signs and symptoms depend on platelet count • onset usually insidious
ITP: Laboratory features • ITP IS A DIAGNOSIS OF EXCLUSION • no sensitive and specific test for ITP • isolated thrombocytopenia • increased MPV • normal PT, PTT • bone marrow investigation not essential in straightforward cases
ITP: TreatmentPatient is not bleeding • plt > 50: Rx not indicated • plt 20-50: Rx usually not needed, monitor closely • plt < 20: Rx indicated with one or more of: • prednisone • IVIG • anti-D if Rh pos • splenectomy if relapsing severe ITP (No role for prophylactic platelet transfusion, even if plt = 0)
ITP: TreatmentPatient is bleeding • For serious bleeding (eg. CNS, retroperitoneal, GI) • Prednisone and IVIG • Transfuse platelets • consider urgent splenectomy • Provide other supportive/resuscitative care as needed
ITP: Prognosis • Children: usually permanent remission • Adults: usually relapsing (chronic ITP), but course is relatively benign.
Features of Acute and Chronic ITP Features Acute ITP Chronic ITP Peak age Children (2-6 yrs) Adults (20-40 yrs) Female:male 1:1 3:1 Antecedent infection Common Rare Onset of symptoms Abrupt Abrupt-indolent Platelet count at presentation <20,000 <50,000 Duration 2-6 weeks Long-term Spontaneous remission Common Uncommon
Incidence(per 105 / year) Age (yrs)Female Male Total 15-39 2.3 1.3 3.6 40-59 3.2 1.1 4.3 60+ 4.6 4.4 9.0 Total 3.2 2.0 2.6 Incidence of adult ITP increases with age
Initial Treatment of ITP Platelet count Symptoms Treatment (per µl) >50,000 None 20-50,000 Not bleeding None Bleeding Glucocorticoids IVIG <20,000 Not bleeding Glucocorticoids Bleeding Glucocorticoids IVIG Hospitalization
Variable No./total (%) Complete response With glucocorticoids 370/1447 (26%) With splenectomy 581/885 (66%) Death from hemorrhage 78/1761 (4%) Healthy at last observation 1027/1606 (64%) Summary of case seriesAdults with ITP
Long-term morbidity and mortalityin adults with ITP • 134 patients with severe ITP studied for mean of 10.5 yrs • CR and PR patients (85%) • No increased mortality compared to control population • Non-responders/maintenance therapy • Increased morbidity due to ITP-related hospitalizations • Increased mortality related equally to bleeding and infection
Approach to the thrombocytopenic patient • History • Is the patient bleeding? • Are there symptoms of a secondary illness? (neoplasm, infection, autoimmune disease) • Is there a history of medications, alcohol use, or recent transfusion? • Are there risk factors for HIV infection? • Is there a family history of thrombocytopenia? • Do the sites of bleeding suggest a platelet defect? • Assess the number and function of platelets • CBC with peripheral smear • Bleeding time or platelet aggregation study
THROMBOCYTOPENIA rule out pseudothrombocytopenia SEQUESTRATION • PRODUCTION • DESTRUCTION Approach to thrombocytopenia look for splenomegaly bone marrow investigation review meds look for underlying disorders review meds • Causes of splenomegaly • infection • inflammation • congestion • maligancy • red cell disorders • storage diseases • aplasia • infiltration • ineffective megakaryopoiesis • eg. MDS • selective impairment of platelet production • immune • auto-immune (ITP, SLE • drugs • infections • allo-immune • non-immune • sepsis • DIC, TTP, HUS • hypertensive disorders of pregnancy
Bleeding time and bleeding • 5-10% of patients have a prolonged bleeding time • Most of the prolonged bleeding times are due to aspirin or drug ingestion • Prolonged bleeding time does not predict excess surgical blood loss • Not recommended for routine testing in preoperative patients
BFU-E/CFU-E CFU-GM CFU-Baso CFU-Eos Thrombocytopoiesis PLURIPOTENT STEM CELL MIXED PROGENITOR CELL COMMITTED PROGENITOR CELL RECOGNIZABLE BONE MARROW PRECURSOR CELL MATURE BLOOD CELL pronormoblast red cell myeloblast monoblast neutrophil monocyte eosinophil myeloid progenitor cell basophil CFU-Meg megakaryocyte platelet pluripotent stem cell pre-T lymphoblast T-cell pre-B lymphoblast B-cell lymphoid & plasma cell
Platelet formation megakaryocyte formation of demarcation membranes platelets (pro)platelets
(+) thrombopoietin Platelets in the circulation:Influx, efflux, and redistribution platelet count CIRCULATING PLATELETS DESTRUCTION or REMOVAL PRODUCTION 70% SPLEEN 30%
normal platelet count thrombocytopenia plasma TPO platelet [TPO] total normal normal [TPO] free normal increased Thrombopoietin
Thrombocytopenic bleeding • Risk of bleeding • platelet count • cause of thrombocytopenia • comorbid disease • drugs • Clinical manifestations • petechiae • purpura, ecchymoses • mucosal bleeding • menorrhagia • intracranial bleeding
Disseminated Intravascular Coagulation (DIC) • DIC is characterized by • the systemic activation of the coagulation system followed by activation of fibrinolytic system • high thrombin and plasmin generation • DIC is not a disease itself, but is a manifestation of a serious underlying disorder.
Causes of DIC • Infection - bacterial sepsis, viral infections • Neoplasm - AML, adenocarcinoma • Obstetrical disorders - retained dead fetus, abruption, etc • Trauma/surgery - brain injury, crush, burns, etc. • Others - acute hemolytic transfusion reaction, etc.
PATHOPHYSIOLOGIC EVENTS LABORATORY MANIFESTATIONS CLINICAL MANIFESTATIONS underlying disorder depletion of clotting factors prolonged PT, PTT tissue factor release thromboctyopenia (consumption) activation of intrinsic pathway of coagulation (systemic thrombin generation) hemorrhage depletion of physiologic anticoagulants decreased fibrinogen generalized intravascular fibrin deposition microangiopathic hemolytic anemia thrombosis/infarction activation of fibrinolytic system (systemic plasmin generation) increased FDP and D-dimer Pathophysiology of DIC
Treatment of DIC • treat the underlying disease • replacement therapy • cryoprecipitate • FFP • platelet concentrate • packed red cells • consider additional pharmacologic therapy • controversial or investigational agents • AT, APC, PC concentrate, heparin, antifibrinolytic agents.
Qualitative Defects in Primary Hemostasis • Adhesion/Adherence Defects • Glycoprotein (Gp) Ib deficiency • Bernard-Soulier syndrome • von Willebrand disease • Aggregation defects • Afibrinogenemia • Release defect • Acquired defects • Antibodies to GP IIb-IIIa in NHL and HD
Aggregation & Release Platelet Function Adherence Only Direction of Blood Flow
von Willebrand Disease vWF Bernard-Soulier Syndrome vWF vWF vWF Platelet Adherence Gp Ib Gp Ib
Wiskott-Aldrich syndrome Hermansky-Pudlak syndrome Gray platelet syndrome Lysosome Chédiak- Higashi anomaly Platelet Release Function ADP ß-thromboglobulin Platelet factor 4 DenseGranule Platelet-derived Growth Factor Alpha-granule Fibrinogen Factor V Hydrolase
vWF Gp Ib IIb IIIa ADP D D Thrombasthenia ADP IIb IIIa D Fibrinogen E IIb IIIa Afibrinogenemia Platelet Aggregation Release Defects Dense Granule
Platelet Aggregation • In-vivo platelet aggregation is induced by • Thrombin mechanisms • Stimulates ADP release • Enhances formation of TxA2 • Thromboxane A2, mediated through • Arachidonic acid release form membrane phospholipid • Cyclo-oxygenase • Endoperoxidase
Laboratory Tests for Primary Hemostasis Function • Platelet count • Bleeding time • Platelet Aggregation Studies • Luminance method • Impedance method • Clot retraction • Not available • Expected results: clots should normally be reduced by 50% of their original mass within 1 hour • Flow cytometric studies for Glycoproteins
Secondary Response % Transmittance Time Platelet Aggregometry Aggregation Plateau Shape Change % Transmittance Reagent Added Note: With impedance method there is increased resistance of the sample and ATP release Baseline Time
Results of platelet aggregation studies • No aggregation with arachidonic acid and ADP • Markedly reduced aggregation with collagen • Normal aggregation with ristocetin • ATP release with thrombin is normal but absent with arachidonic acid
Aggregating Agent Disorder ADP Epi Thr Ris Col Ara Bernard-Soulier syndrome Nor Nor Var Var Nor Chédiak-Higashi anomaly Abn/Var Abn/Var Abn/Var Abn/Var Glanzmann thrombasthenia Abn Abn Abn Abn Granule release defects Abn Abn Abn von Willebrand's disease Abn Platelet Aggregometry interpretation Abn Nor
Flow cytometry for the detection of Gp defeciency • The study of GPIIbIIIa showed reduction in PAC-1 which is a finding consistent with Glanzmann thrombasthenia • However hepatosplenomegaly is not explainable by the diagnosis • Patient was readmitted on 18-11-02 for further investigations to clarify the hepatosplenomegaly
ThrombastheniaSynonyms: Glanzmann thrombasthenia, constitutional thrombopathy, hereditary hemorrhagic thrombopathy • Background: • Thrombasthenia was first describe in 1918 by Glanzmann when he noted purpuric bleeding in patients with normal platelet counts • Typically, thrombasthenia is diagnosed at an early age • Pathophysiology: • Autosomal recessive trait • The production and assembly of the platelet membrane glycoprotein IIb-IIIa is altered, preventing the aggregation of platelets and subsequent clot formation
Review of platelet function • Platelets adhere to the site of endothelial injury • Activate • Aggregate • Secrete & promote further platelet recruitment & aggregation • vWF binds to the exposed collagen and binds GP Ib-IX-V complex on the surface of platelet, adhering platelets to the site of injury • Fibrinogen and vWF bind to the GP IIb-IIIa complex on the activated platelet’s surface, allowing cross-linking and formation of clot
Specific Deficiency • GP IIb and IIIa have separate genes on the long arm of chromosome 17 • Specific genetic abnormalities of each GP include • Missense mutations • Nonsense mutations • Splice site mutations • Deletions and • Point mutations • Abnormalities in either gene or in the assembly of the complex result in an abnormal or deficient receptor
Consequently • One or other GP is not formed properly, leaving the other unpaired in the endoplasmic reticulum, where it is degraded • Platelet aggregation is rendered deficient or completely absent • Heterozygotes are asymptomatic • Binding sites for thrombin are preserved in thrombasthenic platelets
Patients are classified into: type1, type2, or the variant type, depending on the degree of GP IIb-IIIa deficiency, fibrinogen binding, and clot retraction • Type 1 : most severe form, less than 5% of normal GP IIb-IIIa present with absent fibrinogen binding and clot retraction • Type 2 : 10-20% of GP IIb-IIIa, normal to moderately deficient clot retraction with fibrinogen binding • Variant type : 50% of the normal amount of GP IIb-IIIa with extremely variable fibrinogen binding and clot retraction
Frequency: • 300 cases are reported in medical literature • 1st case diagnosed in HUSM • Mortality/Morbidity: • Death following bleeding approx. 5% • Age: • Typically diagnosed during infancy • Epistaxis is more severe in children but rare in adults • History • In the neonatal period: mucocutaneous bleeding • In childhood: purpura, epistaxis & gingival bleeding • The bleeding tendency decreases with age