Download

1 / 32
330 likes | 2.3k Views
Hemolytic anemia. HA = decreased levels of erythrocytes in circulating blood (anemia) because of their acclerated destruction (hemolysis) A red blood cell survives 90 to 120 days (on average) in the circulation, therefore about 1% of human red blood cells break down each day.

E N D
Hemolytic anemia • HA = decreased levels of erythrocytes in circulating blood (anemia) because of their acclerated destruction (hemolysis) • A red blood cell survives 90 to 120 days (on average) in the circulation, therefore about 1% of human red blood cells break down each day. • The spleen (part of the reticulo-endothelial system) is the main organ which removes old and damaged RBCs from the circulation. • In health the breakdown and removal of RBCs from the circulation is matched by the production of new RBCs in the bone marrow. • When the rate of breakdown increases, the body compensates by producing more RBCs, but if compensation is inadequate clinical problems can appear. Breakdown of RBCs can exceed the rate that the body can make RBCs and so anemia can develop. The breakdown products of hemoglobin will accumulate in the blood causing jaundice and be excreted in the urine causing the urine to become dark brown in colour.
HEMOLYTIC ANEMIAHemolytic anemia = reduced red-cell life span
Signs of hemolytic anemia: History • Onset/ duration (hereditary versus acquired) • History of fatigue or jaundice • Abdomen pain / cholelithiasis (chronic hemolysis) • Medications or food /ie fava bean/(may exacerbate enzyme deficiencies) • Travel (consider infection) and infection • Vascular / cardiac surgery • Blood loss or sequestration (increases reticulocytes in the absence of hemolysis) • Discolored urine (intravascular haemolysis) • Complete family history (jaundice, gallbladder disease, splenectomy, hereditary anaemia or other inherited diseases)
Signs of hemolytic anemia: Physical • Symptoms of anemia • Jaundice • Pallor • Splenomegaly / hepatosplenomegaly • Increased temperature • Rapid pulse
Laboratory futures (1) *Morfology: anemia *Peripheral blood smear microscopy: • **fragments of the red blood cells ("schistocytes") can be present • **some red blood cells may appear smaller and rounder than usual (spherocytes) • **reticulocytes are present in elevated numbers. • **erytroblasts can be present. *Bone marrow smear microscopy: erytrhroid hyperplasia (megaloblasts) *The level of unconjugated bilirubin in the blood is elevated. *The level of lactate dehydrogenase (LDH) in the blood is elevated
Laboratory futures (2) *Haptoglobin, hemopexin levels are decreased *Iron level in the blood is elevated (in NNH is decreased!) *The direct Coombs test is positive, if hemolysis is caused by an immune process. *Increased excretion of urobilinogen in the urine and stercobilinogen in the stool. *Sometimes abnormal results of the osmotic fragility test *Free hemoglobin, methemalbumin elevated level in the blood and hemoglobin, hemosiderin in the urine indicates chronic intravascular hemolysis.
Classification of hemolytic anemias (1) • Causes of hemolytic anemis can be either genetic or acquired.
Classification of hemolytic anemias (2)===Genetic=== • *Genetic conditions of RBC membrane • **Hereditary spherocytosis • **Hereditary elliptocytosis • *Genetic conditions of RBC metabolism (enzyme defects) • **Glucose-6-phosphate dehydrogenase deficiency (G6PD or favism) • **Pyruvate kinase deficiency • *Genetic conditions of hemoglobin • **Sickle cell anemia • **Thalassaemia
Classification of hemolytic anemias (3)===Acquired=== '''Immune mediated hemolytic anemia''' (direct Coombs test is positive) • *Autoimmune hemolytic anemia • **Warm antibody autoimmune hemolytic anemia • ***Idiopathic • ***Systemic lupus erythematosus (SLE) • ***Evans' syndrome (antiplatelet antibodies and hemolytic antibodies) • **Cold antibody autoimmune hemolytic anemia • ***Idiopathic cold hemagglutinin syndrome • ***Infectious mononucleosis and mycoplasma ( atypical) pneumonia • ***Paroxysmal cold hemoglobinuria (rare)
Classification of hemolytic anemias (4)===Acquired=== '''Immune mediated hemolytic anemia''' (direct Coombs test is positive) • *Alloimmune hemolytic anemia • **Hemolytic disease of the newborn (HDN) • ***Rh disease (Rh D) • ***ABO hemolytic disease of the newborn • ***Anti-Kell hemolytic disease of the newborn • ***Rhesus c hemolytic disease of the newborn • ***Other blood group incompatibility (RhC, Rhe, RhE, Kidd antigen system, Duffy antigen, MN, P and others) • **Alloimmune hemolytic blood transfusion reactions (ie from a non-compatible blood type) • *Drug induced immune mediated hemolytic anemia • **Penicillin (high dose) • **Methyldopa
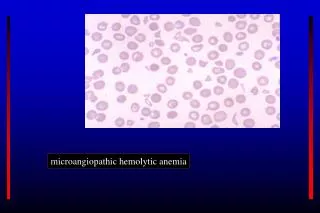
Classification of hemolytic anemias (5)===Acquired=== '''Non-immune mediated haemolytic anaemia''' (direct Coombs test is negative) • *Drugs (i.e., some drugs and other ingested substances lead to hemolysis by direct action on RBCs) • *Toxins (e.g., snake venom) • *Trauma • **Mechanical (heart valves, extensive vascular surgery, microvascular disease) • *Microangiopathic hemolytic anemia (a specific subtype with causes such as TTP, HUS, DIC and HELLP syndrome) • *Infections • **Malaria • **Babesiosis • **Septicaemia • *Membrane disorders • **Paroxysmal nocturnal hemoglobinuria (rare acquired clonal disorder of red blood cell surface proteins) • **Liver disease • *Hypersplenism
Mechanisms of hemolysis:- intravascular - extravascular
Intravascular hemolysis (1):- red cells destruction occurs in vascular space - clinical states associated with Intravascular hemolysis: acute hemolytic transfusion reactions severe and extensive burns paroxysmal nocturnal hemoglobinuria severe microangiopathic hemolysis physical trauma bacterial infections and parasitic infections (sepsis)
Intravascular hemolysis (2):- laboratory signs of intravascular hemolysis: tests for hemolysis and aditionally: hemoglobinemia methemalbuminemia hemoglobinuria hemosiderynuria
Extravascular hemolysis :- red cells destruction occurs in reticuloendothelial system - clinical states associated with extravascular hemolysis : autoimmune hemolysis delayed hemolytic transfusion reactions hemoglobinopathies hereditary spherocytosis hypersplenism hemolysis with liver disease- laboratory signs of extravascular hemolysis: tests for hemolysis
Hemolytic anemia Crises: • hemolytic • aplastic
Differential diagnosis * ''Ineffective hematopoiesis'' is sometimes misdiagnosed as hemolysis. • ** Clinically these conditions may share many features of hemolysis • ** Red cell breakdown occurs before a fully developed red cell is released into the circulation. • ** Examples: myelodysplastic syndrome, megaloblastic anemia.
Therapy Compensated hemolysis – observation (clinical evaluation) and folic acid at an oral dose 1mg/day Decompensated hemolysis (definitive therapy depends on the cause): *Symptomatic treatment -blood transfusion: if there is marked anemia. • *In immune-related hemolytic anemia: steroid therapy , immunosupressive agents, immunoglobulins • *Sometimes splenectomy can be helpful where extravascular heamolysis is predominant (ie most of the red blood cells are being removed by the spleen). • Folic acid Hemochromatosis – chelatic agents (Desferal)
Hereditary microspherocytosis (HS)1. Epidemiology: usually inherited as an autosomal dominant trait; affects about 220 per million people worldwide2. Pathophysiology: red cell membrane protein defects (spectrin, spectrin-ankyrin, bad3 and 4.2 (palladin) deficiency) caused by mutations in he spectrin and ankyrin genes, resulting cytoskeleton instability3. Familly history (at least half of new diagosed patients) 4. Clinical features: jaundice, gallstones, splenomegaly, constitutional skeleton changes (ie tower cranium, gothic palate)5. Laboratory features - hemolytic anemia - blood smear-microspherocytes - abnormal osmotic fragility test, cryohemolysis test, acidified glycerol lysis time - negative direct Coombs test - increased MCHC6. Treatment - splenectomy (patients >6 yrs old) eradicates clinical manifestations of the disorder, including aplastic crises
Paroxysmal nocturnal hemoglobinuria PNH 1. Pathogenesis - an acquired clonal disease, arising from a somatic mutation in a single abnormal stem cell. PNH involves the PIG-A gene (short arm of the X chromsome). The mutation results of glycosyl-phosphatidyl-inositol (GPI) anchor abnormality - deficiency of the GPI anchored membrane proteins (DAF=decay-accelerating factor CD55, MIRL=a membrane inhibitor of reactive lysis, C8BP=C8 binding protein) - the defective synthesis of GPI affects all hematopoietic cells (anemia, neutropenia end thrombocythopenia, or they may have complete BM failure) - red cells are more sensitive to the lytic effect of complement - intravascular hemolysis 2. Symptoms: passage of dark brown urine in the morning, severe pain in he abdomen and recurrent thromboembolism (ie vena cava inf., portal mesenteric system)
3. PNH –laboratory features: - pancytopenia - chronic urinary iron loss - serum iron concentration decreased - hemoglobinuria - hemosiderinuria - positive Ham’s test (acid hemolysis test) - positive sugar-water test - specific immunophenotype of erytrocytes (CD59, CD55)4. Treatment: - washed RBC transfusion - iron therapy - allogenic bone marrow transplantation (streoids may reduce transfusion requirements; splenectomy – very questionable benefit)
SICKLE CELL ANEMIADefinition: chronic hemolytic anemia occuring almost exclusively in blacks and characterized by sickle-shaped red cells(RBCs) caused by homozygous inheritance of Hemoglobin S
SICKLE CELL ANEMIA-pathogenesis - In HbS, valine is substituted for glutamic acid in the sixth amino acid of the ß chain. - Deoxy-HbS is much less soluble than deoxy HbA; it forms a gelatinous network of fibrous polymers that cause RBCs to sickle at sites of low pO2. - Hemolysis - because sickle RBCs are too fragile to withstand the mechanical trauma of circulation - Occlusion in microvascular circulation caused by distorted, inflexible RBCs adhering to vascular endothelium
SICKLE CELL ANEMIA-incidence - Homozygous - about 0.3% of blacks in the USA (have sickle cell anemia) - Heterozygotes-8-13% of blacks, (are not anemic, but the sickling trait=sicklemia can be demonstrated in vitro)
SICKLE CELL ANEMIA-clinical features IN HOMOZYGOTES 1. Clinical complications due to severe hemolytic anaemia - slowed growth and development in children - bilirubins stones - aplastic crisis - congestive heart failure from chronic anemias and cardiac overload compensation 2. Consequences of vaso-occlusion of the microcirculations (tissue ischemia and infarction) - infarction of spleen, brain, marrow, kidney, lung, aseptic necrosis, central nervous system and ophtalmic vascular lesions
SICKLE CELL ANEMIA-laboratory findinges 1. Anemia-normocytic or slightly macrocytic 2. Leukocytosis (chronic neutrophilia) 3. Thrombocytosis - usually mild<1000G/l 4. Reticulocytosis 5. Peripheral smear: sickle shaped red cells, polychromatophilia, Howell-Jolly bodies 6. Hb –electrophoresis or high-performance liquid chromatography (HPLC)
SICKLE CELL ANEMIA-therapy Preventive measures: prevention or remedy of: infections (penicillin prophylaxis and pneumococcal vaccination), fever, dehydratation, acidosis, hypoxemia, cold exposure, pain Blood transfusions for very severe anemia New approaches to therapy; 1. Activation of HbF synthesis: hydroxyurea, 5-azacytidine, decytabine 2. Antisickling agents acting on hemoglobin or membrane (preclinical testing, clinical trials) 3. Bone marrow transplantation 4. Gene therapy
Thalasemias • The regions in which thalasemia occure are contiguous with regions endemic for malaria (protection against malaria) • Thalasemia result from gene (located on chromosomes 11 and 16) deletion, abnormalities in transcription and translation and instability of the mRNA directing globin synthesis or of the globin itself. • Result: imbalanced synthesis of normal globin chain. The unpaired chain accumulates in the developing erythroid precursor cell, and toxicity results – ineffective erythropoiesis, hemolysis and anemia of variable degree.
Different forms of thalassemia • Alfa thalassemia • Beta thalasemia: major, minor (trait), intermedia • Delta/Beta thalassemia • Hereditary persistentce of fetal hemoglobin (HPFH)
Beta-Thalassemia major (Cooley anemia) • Usually homozygous condition • In the most severe variant no beta-chains are synthesized • Clinical futures: sever anemia that appears in the first year of life; jaundice, hepatosplenomegaly (secondary neutropenia and thrombocytopenia), skin pigmentation and chronic leg ulceration, expansions of the erythroid marrow with secondary body changes (including retarded growth, bossing of skull, expanded maxilla, widened diploe, gross skeletal deformities, spontaneous fractures, dental problem), increased susceptibility to infection, symptoms of iron overloading
Beta-Thalassemia major laboratory features • Severe anemia • Blood film: anisopoikilocytosis, hypochromia, target cells, basophylic stippling, reticulocytes – moderate increased • Marrow: marked erythroid hyperplasia, increased sideroblasts • Shortened red cell survival • Fetal hemoglobin > 90%, HbA absent, HbA2 low/normal/high
Beta-Thalassemia major treatment - High standard of pediatric care required!!! (early treatment of infections, vaccination, folate supplemets, dental care), replacement therapy and chelation when iron-overloading • Transfusion (Hb must be 10 to 14 g/dL) each 6-8 weeks • Splenectomy - BMT - Experimental therapy: hydroxyurea, somatic gene therapy