Download

1 / 32
320 likes | 803 Views
Errores Innatos Del Metabolismo Y Sindrome Parkinsoniano. Francisca Montoya Salvadores Residente de Neurología Universidad de los Andes. Errores innatos del metabolismo. Término acuñado por Archibald Garrod , 1927 Hasta hace poco, limitado a pediatría: RN e infancia

E N D
Errores Innatos Del Metabolismo Y Sindrome Parkinsoniano Francisca Montoya Salvadores Residente de Neurología Universidad de los Andes
Errores innatos del metabolismo • Término acuñado por ArchibaldGarrod, 1927 • Hasta hace poco, limitado a pediatría: RN e infancia • Trastornos monogénicos producen la alteración de una enzima • Trastorno en la síntesis o catabolismo de proteínas carbohidratos o lípidos • Disrupción de las vías metabólicas • Acumulación tóxica de sustratos, metabolitos intermedios de vías alternativas y/o defectos en la producción y/o utilización de energía
Clasificaciones • Compromiso de cualquier órgano o sistema • Perfil temporal: agudo-grave vs. subagudo, progresivo degenerativo • Todos pueden aparecer en adultos
Interfiere con la embriogénesis: dismorfias, displasia y malformaciones • Adultos con encefalopatía: • Intoxicación: RM N: • Ciclo de la urea, porfirias, defectos de metilación de homocisteína • Metabolismo energético: RM alt: • Trastorno de cadena respiratoria, deficiencia de piruvato deshidrogenasa, biotina Inicio adultez ( >70 ) Afecta organelos: lisosomas, peroxisomas, defectos de síntesis de colesterol y glicosilación Presentación psiquiátrica o neurológica Psicosis atípica, depresión, coma, NP periférica, ataxia cerebelosa, paraparesia espástica, demencia, epilepsia y trastornos del movimiento No interfiere con el desarrollo embrionario: intervalo libre Ataque metabólico agudo Crónico
Errores innatos del metabolismo y trastornos del movimiento • Se pueden presentar en la adolescencia o adultez • Trastornos del movimiento asociados: • PKS, distonía, corea, tics, mioclonus • Causas determinadas por la sensibilidad de los GB a: • Enfermedades por depósito de metales • Trastornos del metabolismo energético • Trastornos por depósito lisosomal • Defectos en la síntesis de NT*
Enfermedad de Gaucher • Trastorno lisosomal AR: deficiencia de la glucocerebrosidasa • Síntomas Neurológicos: • Epilepsia mioclónica progresiva, PKS, oftalmoplejia horizontal supranuclear, ataxia, • Otros síntomas: • Astenia, hepatoesplenomegalia, trombopenia, anemia, manifestaciones óseas • Exámenes: • de la actividad de la glucocerebrosidasa: en leu periféricos o cultivos de fibroblastos en piel biopsiada • Células de Gaucher en bx MO: MQ grandes, cargados de lípidos con un nu excéntrico e inclusiones fibrilares citoplasmáticas levemente basófilas • Genético: Gen Glucocerebrosidasa: cromosoma 1q21
Fisiopatología Estado heterocigoto de mutación GBA en PKS y DCL: rol patógeno más allá de la deficiencia enzimática Ubicuitina-Proteosoma
Tratamientos • Tx - esplenectomía y cx ortopédica Trasplante alogénico MO (1980) • Terapia de reemplazo hormonal • 3 productos con eficacia en parámetros hematológicos y vicerales • Terapia de reducción de sustratos • Miglustat: Inhibe la síntesis de glucosilceramide • Única que atraviesa la BHE • Chaperonas • Mutaciones patogénicas en el gen GBA llevan a mal plegamiento proteico y degradación prematura • Chaperonas se unen a las proteínas en el retículo endoplásmico, las estabilizan y ayudan a mantener la correcta conformación
Defectos en la síntesis de las monoaminas • Defectos en las vías de biosíntesis de aminas biogénicas • Estudio: medición directa de NT en LCR • Salvo la deficiencia de la GTP ciclohidrolasa I • Hiperprolactinemia indica defecto en la síntesis de dopamina • Ejemplos • Enfermedad de Segawa: deficiencia de la ciclohidrolasaI, AD mutación gen GTPCH1 • Se presenta a cualquier edad con distoníao PKS, paraparesiapseudoespástica, fluctuaciones diurnas • Exámenes: biopterinas, neopterinas HVA y 5-HIAA en LCR N o . Test de carga de Fenilalanina anormal • Tratamiento: Levodopa, anticolinesterásicos, agonistas dopaminérgicos
Defectos en la síntesis de las monoaminas • Ejemplos • Deficiencia de tirosina hidrolasa, AR • Síntomas: distoníao PKS con poca o nula respuesta a L-dopa, paraparesiapseudoespástica, signos piramidales y RM • Exámenes: HVA en LCR con biopterinas, neopterinas y 5HIAA Ns • Tratamiento: Levodopa, anticolinesterásicos, agonistas dopaminérgicos • Deficiencia de dihidropteridinareductasa, AR • Síntomas: RM, distonia, PKS, epilepsia, depresión • Exámenes: hiperfenilalaninemia, biopterinas,HVA y 5HIAA en LCR • Tratamiento: Levodopa, 5HTP, dieta baja en fenilalanina
Hiperfenilalaninemia • Trastorno intermediario del metabolismo, AR • Síntomas neurológicos • PKS, atrofia óptica, demencia, leucoencefalopatía • Exámenes: hiperfenilalaninemia, hipotirosinemia • Tratamiento: Dieta baja en fenilalanina
Xantomatosis cerebrotendínea • Trastorno del metabolismo intermediario AR • Síntomas neurológicos: • Ataxia cerebelosa, PKS, paraparesia espástica, trastornos psiquiátricos • Otros síntomas: • Cataratas juveniles, xantomas tendíneos, diarrea • RM: hiperintensidad en nucleo dentado • Exámenes: colestanol elevado • Tratamiento: Ácido quenodeoxycólico
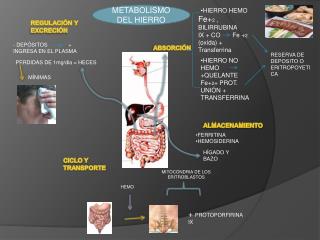
Hemocromatosis • Enfermedad de depósito AR – mut C282Y • Síntomas Neurológicos: • PKS, ataxia cerebelosa, demencia, temblor de intención, mioclonias, distonía, sd piramidal • Síntomas Sistémicos: • Trastornos hepáticos, artritis, cardiomiopatía, coloración bronceada de la piel, astenia • RM: N o atrofia cerebral • Exámenes: Fe sérico alto, saturación de la transferrinay feritina. • Tratamiento: • Flebotomía (no eficiente en PKS) • PKS puede o no responder a L-dopa,
Trastornos de la cadena respiratoria • Trastorno del metabolismo energético • Cualquier tipo de herencia: buscar herencia mitocondrial o mutaciones del DNA nuclear específicas • Síntomas Neurológicos • PKS, distonía, mioclonias • Otros síntomas dependen del sd (oftalmoplejia externa, PNP, endocrinopatía, retinitis pigmentosa) • Exámenes: aumento del lactato en LCR • Bx: fibras rojas rasgadas • Tratamiento: sintomático
Lipofuscinosisceroidea neuronal • Trastorno neurodegenerativo progresivo en niños secundario a acumulaciónde lipopigmentosautofluorescentes en las neuronas y glias • Herencia AR • Clínica: Epimioclónica progresiva asociada a trastornos visuales • 4 formas: Infantil (1,5 ±0,5á), infantil tardío (3,5 ±0,5á), Juvenil (6,5 1,8±á) Adulto (44 ±05,6) • Regresión de los hitos del DSM, convulsiones, mioclonias, corea, amaurosis y ataxia • Adultos: Trastorno de comportamiento y signos extrapiramidales • Muerte 5 a 10-12 á en las formas infantiles y a los 15-25á en las juveniles. Adulto tiene curso variable
Lipofuscinosisceroidea neuronal • Exámenes • EEG: enlentecimiento difuso y generalizado, con descargas epilépticas en 81% • Otros: SSEP gigantes, VEP P100 prolongados, EMG: neuropatía axonal; atrofia difusa en neuroimágenes • Bx: Cerebro, piel músculo hígado • Cerebro: atrofia del manto cortical, deplesión neuronal y presencia de astrocios reactivos. Tinción PAS y luxolfast-blue revelan inclusiones intracitoplasmáticas intensas, granulares. • Microscopio luz fluorescente: sustancia amarilla autofluorescente • Bx piel y músculo son Ns en microscopá de luz. Estudios ultraestructurales muestran las inclusiones
Niemann-Pick C disease • Trastorno lisosomal, AR • Se presenta a cualquier edad: depósito de esfingomielina • Clínica: mioclonias, distonía, corea, PKS, trastornos psiquiátricos, parálisis supranuclear de la mirada vertical, ataxia cerebelosa, demencia • Otros: hepatoesplenomegalia
Deficiencia primaria de la actividad ácida de la esfingomielinasa (A y B) Deficiencia del procesamiento celular y del transporte de colesterol LDL
Niemann-Pick C disease • Exámenes: tinción filipina anormal en fibroblastos • Se reduce la esterificación y se produce una excesiva acumulación de colesterol • MRI: N o atrofia cerebelosa • Tratamiento: Miglustat: inhibidor de la biosíntesis de glicoesfingolípidos • Reduce el depósito de lípidos, mejora la captación endosómicay normaliza el tráfico de lípidos en los linfocitos B • Datos limitados de que su uso puedan prevenir o reducir la progresión de la enfermedad
Enfermedad de Wilson • Trastorno metabolismo de metales, AR, mutación del gen ATP7b: transportador Cu-ATPasa – Acumulación de Cu en cerebro (GB) e hígado • Ceruloplasmina y ferritina juegan un rol crítico en el metabolismo de metales en el cerebro • Ceruloplasmina: actividad ferroxidasa para liberar el Fe de su sitio de almacenaje • Ferritina: fuente de Fe intracelular • Cu es un cofactor que interviene en la movilización del Fe • Cu se fija a la apoceruloplasmina, le da estabilidad y evita su degradación. • Clínica: • Adultos jóvenes 7ma década • Temblor, PKS, distonía, corea, signos psiquiátricos, disartria
Enfermedad de Wilson • Otros: anillos de kayser-Fleischer, DHC • Exámenes: Cu urinario , Cu sérico y ceruloplasmina • RM: depósitos de metales en GB • Atrofia cerebral • T1 en nulentiforme y mesencéfalo en estadio inicial • T2 en globo pálido • T2 caudado, putamen, tálamo VL , nu dentado y puente • Tratamiento: D-penicilamina, zinc, trientina
Neurodegeneración asociada a la pantotenatokinasa: Hallervorden-Spatz • Trastorno metabólico metales, AR, mutación gen PANK2 • Pank2 cataliza la síntesis de coenzima A desde vitamina B5 • Presenta desde la primera década • Clínica: PKS, distonía, disartria, trastornos psiquiátricos y cognitivos, signos piramidales, acantocitosis (8-10%) retinitis pigmentosa • RM: signo de los ojos de tigre • T2 en el globo pálido con un central • T2 GRE: á de susceptibilidad magnética • Manejo: sintomático
Conclusión • Trastornos metabólicos son poco frecuentes de forma aislada • Algunos se pueden presentar con trastornos del movimiento: diagnóstico diferencial parkinsonismos • Alto nivel de sospecha • Importancia: algunas son tratables y definir pronóstico