Download

1 / 24
240 likes | 349 Views
Beyond Teraflops toward Petaflops Computational Chemistry: Challenges and Opportunities. Martyn F. Guest and Paul Sherwood CCLRC Daresbury Laboratory m.f.guest@daresbury.ac.uk. Outline. From Gigaflops to Teraflops - Computational Chemistry Today Migration from replicated to distributed data

E N D
Beyond Teraflops toward PetaflopsComputational Chemistry:Challenges and Opportunities Martyn F. Guest and Paul Sherwood CCLRC Daresbury Laboratory m.f.guest@daresbury.ac.uk 1
Outline • From Gigaflops to Teraflops - Computational Chemistry Today • Migration from replicated to distributed data • Parallel linear algebra (diagonalisation, FFT etc.) • Exploiting multiple length and time scales • From Teraflops to Petaflops • Problem scaling and re-formulation - Four Dimensions • Time to Solution, Problem Size, Enhanced Sampling, Accuracy • Long Term - New horizons for simulation • “Simulation of whole systems, and not just system components” • Scientific Challenges in Key Application areas • Catalytic Processes, Biomolecular Simulations, Heavy Particle Dynamics • “Current Status”, “Towards Petaflops”, “New Horizons” • The Software Challenge • Recommendations and Summary 2
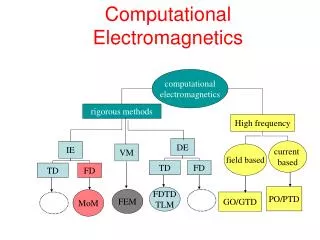
Distributed Memory 10TF NAMD 1TF NWChem GAMESS-UK DL_POLY 3 Shared Memory CHARMM 100GF Gaussian 10GF How Today’s codes exploit Today’s Hardware HPCx 3
High-End Computational ChemistryThe NWChem Software • Capabilities (Direct, Semi-direct and conventional): • RHF, UHF, ROHF using up to 10,000 basis functions; analytic 1st and 2nd derivatives. • DFT with a wide variety of local and non-local XC potentials, using up to 10,000 basis functions; analytic 1st and 2nd derivatives. • CASSCF; analytic 1st and numerical 2nd derivatives. • Semi-direct and RI-based MP2 calculations for RHF and UHF wave functions using up to 3,000 basis functions; analytic 1st derivatives and numerical 2nd derivatives. • Coupled cluster, CCSD and CCSD(T) using up to 3,000 basis functions; numerical 1st and 2nd derivatives of the CC energy. • Classical molecular dynamicsand free energy simulations with the forces obtainable from a variety of sources 4
Time (sec) Number of processors Memory-driven Approaches: NWChem - DFT (LDA) Performance on the SGI Origin 3800 Zeolite ZSM-5 • DZVP Basis (DZV_A2) and Dgauss A1_DFT Fitting basis: • AO basis: 3554 • CD basis: 12713 • MIPS R14k-500 CPUs (Teras) • Wall time (13 SCF iterations): • 64 CPUs = 5,242 seconds • 128 CPUs= 3,451 seconds • Est. time on 32 CPUs = 40,000 secs • 3-centre 2e-integrals = 1.00 X 10 12 • Schwarz screening = 5.95 X 10 10 • % 3c 2e-ints. In core = 100% 5
Multiple Time and Length Scales • QM/MM - first step towards multiple length scales • QM treatment of the active site • reacting centre • problem structures (e.g. transition metal centres) • excited state processes (e.g. spectroscopy) • Classical MM treatment of environment • enzyme structure, zeolite framework, explicitand/or dielectric solvent models • Multiple time scale algorithms for MD • Recompute different parts of energy expression at different intervals e.g. variants of the Reference System Propagation Algorithm (RESPA) But to date length / time scales only differ by ~ 1 order of magnitude For an example of an effort to link the atomistic and meso-scales see RealityGrid: http://www.realitygrid.org/information.html 6
T 128 (O3800/R14k-500) = 181 secs Measured Time (seconds) QM/MM Applications • Triosephosphate isomerase (TIM) • Central reaction in glycolysis, catalytic interconversion ofDHAP to GAP • Demonstration case within QUASI (Partners UZH, and BASF) • QM region 35 atoms (DFT BLYP) • include residues with possible proton donor/acceptor roles • GAMESS-UK, MNDO, TURBOMOLE • MM region (4,180 atoms + 2 link) • CHARMM force-field, implemented in CHARMM, DL_POLY 7
From Teraflops to Petaflops • Short Term - Problem Scaling and Re-formulation • Approaches to efficient exploitation of larger systems • Opportunities for more realistic modelling • Need to avoid dependency on continued scaling of existing algorithms • Scientific Targets: Catalysis, Enzymes and Biomolecules, Heavy Particle Dynamics • Long Term - New Horizons • “Simulation of whole systems, and not just system components” • Automated problem solution • Focus on parallel supercomputers build with commodity compute servers tied by high performance communication fabric • New PetaOPs architecture Projects • IBM’s Blue Light • cellular architecture with 105 or more CPUs; intended to be general purpose • IBM’s Blue Gene • collocation of 32 CPUs, 8MB RAM on same chip (may scale to 106 CPUs) • application specific, protein folding 8
Accuracy • Same model but exploit faster execution • Longer timescale for simulations • Interactive exploration! Limited scalability of current algorithms! Long timescales demand higher accuracy • More Accurate Methods • Better forcefields • Increased use of ab-initio methods • Higher level QM • QM/MM, DFT replacing semi-empirical methods • Finer numerical grids • !Accuracy of methods tends to increase slowly with cost • !Most major challenges involve larger systems • Larger problems • Distributed data methods can exploit large global memories • !Many algorithms contain serious bottlenecks, e.g. diagonalisation O(N3) • !Sampling confor-mational space becomes harder with system size Time “State of the Art” Tera-flop computing ~ 1000 Processors Size Sampling • Study many configurations or systems at once • Better Statistics, Free energies • Combinatorial methods • MC Ensembles!Often satisfied by cheaper, commodity systems 9
What is Petaflop Computational Chemistry • Consider large classical simulations • start from a typical 100,000 particle biomolecule + water • cost per timestep ~50 seconds on 1GF (peak) processor • MD simulations of order 0.5s will require approx 100,000,000 steps • Cost 5,000 Pflops per state point i.e. 1.5 hours on a Pflop machine • Energy Scales as scales as O(N log (N)) and the equilibration time as O(N 5/3) • 1,000,000 particle simulation (for 25 us) will take 840 Pflop hour (complex membrane protein) • Quantum Simulations • Assuming a cost of 5 hours on 1 Gflop processor, 1 day on a 1 Pflop resource will simulate 25 nanoseconds of motion, corresponding to the equilibration time of a 10,000 atom system. 10
Modelling of Catalysis 1. Current Status • Tools • Classical simulation • equilibrium structures and transport properties • QM simulations • reactivity of molecular models, surface structures, supercells • QM/MM methods • solvent, ligand and lattice effects on local chemistry • Scientific Drivers • From mechanisms to reaction rates • From simplified models to multi-component systems, defect sites Collaboration QUASI (Quantum Simulation in Industry) see http://www.cse.clrc.ac.uk/qcg/quasi 11
Accuracy Time Size Sampling Modelling of Catalysis2. Towards Petaflops • Extended dynamical simulation, solvent, counterions etc • Advanced forcefields (polarisabilities, cross-terms etc) • More extensive use of quantum methods (large DFT clusters and periodic supercells) • Combinatorial approach to catalyst formulation • Quantum dynamics & tunnelling via path integral methods • Finite difference approach to local surface vibrations • ab-initio treatment of larger surface domains 12
Modelling of Catalysis3. New Horizons Require integrated models spanning time and length scales. • Initially the interface between the length and time scales will be via construction of parametric models • There is also the possibility that lower-level data might be computed on demand as required to sustain the accuracy of the large-scale simulation Reactor geometries Diffusion of reactants and products Heterogeneous surface structures (films etc) Reaction rates Detailed chemical energetics 13
Biomolecular Simulation 1. Current Status • Tools • Classical Simulation • Simple but well established forcefields for proteins, nucleic acids, polysacharrides etc • QM and QM/MM • enzyme reaction energetics, ligand binding • Continuum electrostatics • Poisson-Boltzmann, Generalised Born • Statistical Mechanics • Scientific Targets • Accurate free energies for more complex systems • Faster and more accurate screening of protein / ligand binding • Membrane proteins (e.g. receptors) • Complex conformational changes (e.g. protein folding) • Excited state dynamics 14
Accuracy Time Size Sampling Biomolecular Simulation2. Towards Petaflops • Faster simulations to approach real timescales • !! major scalability problems • New forcefields incorporating polarisation, cross-terms, etc • Increased use of ab-initio methods • Tremendous potential due to importance of free energies. • Multiple independent simulations • Replica path - simultaneous minimisation or simulations of an entire reaction pathway • Replica exchange - Monte Carlo exchange of configurations between an ensemble of replicas at different temperatures • Combinatorial approach to ligand binding • membranes, molecular assemblies … 15
P0 P1 P2 P3 P4 P32 P33 P34 P35 P36 E Reaction Co-ordinate Replica Path Methods • Replica path method - simultaneously optimise a series of points defining a reaction path or conformational change, subject to path constraints. • Suitable for QM and QM/MM Hamiltonians • Parallelisation per point • Communication is limited to adjacent points on the path - global sum of energy function Collaboration with Bernie Brooks (NIH) http://www.cse.clrc.ac.uk/qcg/chmguk 16
Biomolecular Simulation 3. New Horizons • Towards full quantum simulation (e.g. Car Parrinello) • Towards Whole cell simulation • Mechanical deformation, Electrical behaviour • Diffusion of polymeric molecules (e.g. neuro-transmitters) by DPD • Nanoscale models for supra-molecular structures (e.g. actin filaments in muscles) • Atomistic Molecular Dynamics • Quantum chemistry of reacting sites CCP1 Flagship project - Simulation of Condensed Phase Reativity: http://www.ccp1.ac.uk/projects.shtml 17
Heavy Particle Dynamics 1. Current Status • Tools • Many methods require evaluation of energies on a massive multi-dimensional grid • Use high-level computational chemistry methods (e.g. NWChem, MOLPRO), together with task farming. • Complex parameter fitting • Can exploit interactivity (INOLLS) incorporating experimental data • Dynamical Simulation Methods • Wavepacket evolution on a Grid • Classical path methods (multiple direct dynamics trajectories) • Variational solutions for spectroscopic levels • Targets • Larger species (5 atoms and beyond) • Influence of surrounding molecules (master equation) CCP6 Collaboration, ChemReact consortium on national HPC facilities 18
Accuracy Time Size Sampling Heavy Particle Dynamics2. Towards Petaflops • Interactivity in potential energy surface fitting. • Longer time simulations (slower reactions) • PE surfaces from large basis set CCSD(T) etc • MRCI for excited states and couplings • Large grids for higher dimensionality systems (5 atoms) • Multiple coupled PE surfaces (involvement of excited states) • J > 0 - additional angular momentum states • Larger grids for wavepacket evolution 19
Heavy Particle Dynamics3. New Horizons • Integration with Combustion, detonation, atmospheric models, most likely through detailed parameter and rate constant derivation CFD simulation (transient or steady state, turbulence models etc) Reduced chemical kinetics Reaction rates, sensitivity to pressure and temperature Heavy particle dynamics 20
The Software Challenge • Lack of sustained focus within the Chemical Sciences for responding to the challenges of Petascale Computing • The impact of providing such a focus periodically demonstrated at points on the road to Terascale Computing • The HPCC Grand Challenge projects • NWChem - DOE (PNNL) - principally Electronic Structure; • NAMD - NIH funded initiative in bio-molecular sciences and Classical MD. • Such Initiatives demand: • The successful integration of multi-disciplinary teams including Application and Computational scientists, Computer Scientists and Mathematicians; • A long term commitment to the challenge, with funding in place to respond to the inevitable pace of architecture / hardware change. • Relying on the efforts of individual groups to overcome this software challenge will not work. 21
Problem Solving Environments Requirement:A comprehensive problem solving environment (PSE) for molecular modeling and simulation. Key components include: • common graphical user interfaces • scientific modelling management • seamless transfer of information between applications • persistent data storage • integrated scientific data management • tools for ensuring efficient use of computing resources across a distributed network i.e. GRID • visualization of multi-dimensional data structures 22
Summary and Recommendations 1. Computational chemistry on Tera-scale resources “needs work”, but there are plenty of opportunities to advance collaboratively chemical sciences “towards petaflops” • The short term priority is scaling and adapting current methodologies • Advancing use of distributed data algorithms • O(N) techniques to remove bottlenecks and enhance scalability • Detailed work on parallel scaling • Library developments • Performance analysis and prediction tools • Re-formulation of problems in terms of more weakly interacting ensembles • Parallel implementations of more complex physical models • Automation, data handling, PSEs for combinatorial work • In the longer term by tackling more integrated problems • Modularity of software • Science of the time / length scale interfaces 23
Summary and Recommendations 2. • Investment in Software: “Code Sharing” • UK has kept a strong applications focus, but has lagged behind the US in the radical re-design of simulation packages • Sustained investment in Petascale Software Development • Current UK and International collaborations • Scalable QC algorithms • NWChem, MOLPRO, GAMESS-UK (PNNL, ORNL, SDSC, DL, CCP1/5/6); • Replica path methods in CHARMM/GAMESS-UK • DL collaboration with NIH, PSC; • Flexible QM/MM models incorporating classical polarisation • ChemShell / GULP / GAMESS-UK / NAMD; • Distributed data classical models, electrostatic models • DL_POLY, NWChem – CCP1/5. • Stronger Links between UK initiatives and US programs SciDAC and NSF PACI, NPACI etc. 24