Download
1 / 12
120 likes | 233 Views
Trinuclear Mixed-Valent Oxo-Centered Iron Complexes: Fully Localised, Partially and Fully Delocalised Valencies. R.W. Saalfrank, A. Scheurer, Universität Erlangen-Nürnberg, Germany V. Schünemann, Technische Universität Kaiserslautern, Germany

E N D
Trinuclear Mixed-Valent Oxo-Centered Iron Complexes: Fully Localised, Partially and Fully Delocalised Valencies R.W. Saalfrank, A. Scheurer, Universität Erlangen-Nürnberg, Germany V. Schünemann, Technische Universität Kaiserslautern, Germany A.X. Trautwein, Universität zu Lübeck, Germany
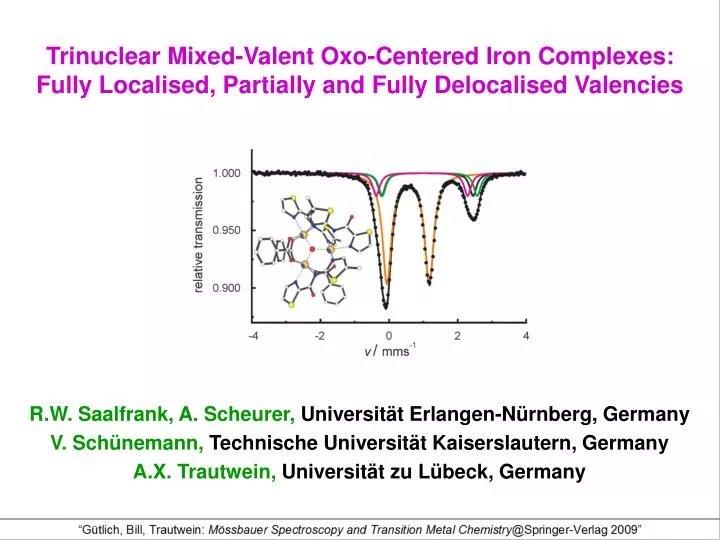
Motivation Our interest in polynuclear supramolecular {Fe3O} complexes stems from the importance of oxo-centered polyiron aggregates as model compounds for iron-oxo proteins, oxidation catalysts, and corrosion inhibitors. Such oxo-centered complexes, related to basic iron acetate [FeIII3O(O2C-R)6(H2O)3]+, are easily accessible (Fig. 1, left): Deprotonation of pentadentate ligand H2L1 with triethylamine and treatment of the resulting dianion L2- with a solution of iron(III) chloride, yielded after workup the triple-helical, μ3-oxo-centered, trinuclear and mixed-valent complex [FeIIFeIII2O(L1)3] (2) as dark-green crystals (Fig. 1, right: stereoview) [1]. Stereoview of 2: FeII dark blue, FeIII gold, C white, N blue, O red Fig. 1
The lack of counterion in the solid state implies intramolecular charge compensation and therefore mixed-valence character for [FeIIFeIII2O(L1)3] (2). This is confirmed by a Mössbauer spectrum of a powder sample recorded at 4.2K. The spectrum exhibits two quadrupolar doublets with a peak area ratio of about 1:2 (Fig. 2). The doublet with quadrupole splitting DEQ=2.64 mms-1 and an isomer shift d=0.95 mms-1 suggests a high-spin iron(II) species and the doublet with DEQ=1.83 mms-1 and d=0.53 mms-1 a high-spin iron(III) species. From this view 2 appears as a localised mixed-valent trinuclear complex with ground state of the type Fe2+-Fe3+-Fe3+ as observed also in other cases [2]. A more rigorous characterisation of the valencies in 2, however, requires a more detailed investigation (vide infra), including the variation of temperature. Fig. 2
Stereoview of 3, Fe gold, C white, N blue, O red, S yellow Fully localised vs. fully delocalised valency The complex [Fe3O(OAc)6(3-Et-py)3].CH3CCl3 (3), with 3-Et-py representing 3-ethylpyridine (Fig. 3, left), exhibits dI=0.54 mms-1, DEQI=1.04 mms-1 for the ferric and dII=1.26 mms-1, DEQII=1.54 mms-1 for the ferrous site at T=43 K [3]. At room temperature only one doublet with d =0.65 mms-1 and DEQ=0.93 mms-1 is seen (Fig. 3, right); this corresponds to full delocalisation of the “excess” electron over the three iron sites. If the solvate molecule CH3CCl3 is replaced by toluene or benzene, however, the complex remains valence trapped up to room temperature. Fig. 3 (Mössbauer spectra taken from [3])
Partially valence delocalised states The Mössbauer spectra of [FeIIFeIII2O(L1)3] (2) in the temperature range between 4.2 and 332 K are shown in Fig. 4 [4]. A consistent analysis of all spectra requires four doublets. They are pairwise related by the area ratio I:II and III:IV of 1:2. From dI and dII2 appears as mixed-valent iron complex which exhibits a partially delocalised ground state: Fe2.2+-Fe2.9+-Fe2.9+ (vide infra). With increasing temperature the subspectra III and IV are gaining weight. They represent Fe2.5+-Fe2.75+-Fe2.75+, a structurally slightly different molecular state of [FeIIFeIII2O(L1)3] (2) in the solid. The two complexes Fe2.2+-Fe2.9+-Fe2.9+ and Fe2.5+-Fe2.75+-Fe2.75+ do not significantly change their respective delocalisation pattern of the “excess” electron, however, their relative amounts do change with increasing temperature: at 4.2 K the relative amounts are 90 % and 10 %, and at 332 K they are 45 % and 55 %. I II III IV Fig. 4
Fig. 4: The spectrum taken at 4.2 K is dominated by two doublets, I and II, with intensity ratio 1:2 and with the isomer shifts and quadrupole splittings of dI=0.99 mms-1, DEQI=2.65 mms-1 and dII=0.52 mms-1, DEQII=1.85 mms-1. It has been found in an accompanying study that the isomer shift of the ferric sites of [FeIIFeIII2O(L1)3] (2) (d=0.52 mms-1) is enhanced compared to the value of the ferric sites of the corresponding hetero-nuclear complexes [MIIFeIII2O(L1)3] with MII=NiII, CoII and CuII, i.e. d=0.46 mms-1. In turn, the isomer shift of the ferrous site (d=0.99 mms-1) is significantly lower than the corresponding d-values expected for 5N/1O ligation; e.g. isomer shifts of various octahedrally coordinated iron(II)-pyridine complexes [FeII(pyridine)4X2] with X=Cl, Br, I, NCO are ~1.1 mms-1 at 4.2 K. Both the enhancement of the isomer shift of the ferric sites as well as the reduction of the isomer shift of the ferrous site can be explained on the basis of partial electron (valence) delocalisation in the ground state of [FeIIFeIII2O(L1)3] (2) according to the relation d,Fe(2+x)+ = xd,Fe2+ + (1-x)d,Fe3+ d,Fe(3-y)+ = yd,Fe2+ + (1-y)d,Fe3+ and x = 2y. From this it has been concluded that 2exhibits a partially delocalised ground state at 4.2 K, i.e. Fe2.2+-Fe2.9+-Fe2.9+. A corresponding estimate with subspectra III and IV (dIII=0.80 mms-1 and dIV=0.60 mms-1 at 20K) yields Fe2.5+-Fe2.75+-Fe2.75+ [4]. The main contribution to the observed temperature dependence of isomer shifts in Fig. 4 is due to the second-order Doppler shift (SOD). For comparison, SOD accounts for Dd ~ 0.13 mms-1 in the temperature interval between 4.2 and 300 K for [FeII(pyridine)4X2] complexes [5]. If the temperature dependence of isomer shifts in 2were mainly due to the temperature-induced changes of electron delocalisation, then the isomer shifts dI and dII as well as dIII and dIV would gradually merge pairwise with increasing temperature according to the above relation. Such behavior was not observed in the present case, therefore it was concluded that the clusters Fe2.2+-Fe2.9+-Fe2.9+ and Fe2.5+-Fe2.75+-Fe2.75+ do not change their respective delocalisation pattern of valencies.
Solvent vs. packing effect upon valence delocalisation Electron (valence) delocalisation is very much affected by solvent and packing. Fig. 4 has shown the temperature-dependent Mössbauer spectra of [FeIIFeIII2O(L1)3] (2) in the solid state with partially delocalised valencies over the whole temperature range. Fig. 5 dis- plays the Mössbauer spectra of 2 at 4.2K (a) and at 160 K (b) in frozen THF [6]. Only three doublets are required to fit the spectra at 160 K. Two of them (I and II) are consistent with partially localised valencies, i.e. Fe2.3+-Fe2.85+-Fe2.85+, while the third (III; d=0.62 mms-1) is consistent with full valence delocalisation, i.e. Fe2.67+-Fe2.67+-Fe2.67+. This apparently delocalised component accounts for as much as 70% of the molecules at 160 K. Fig. 5
Electron exchange in asymmetrically substituted hetero-nuclear {MIIFeIII2O} complexes The oxo-centered iron(II,III) complex [FeIIFeIII2O(L2)3(OAc)3] (5) was synthesised starting from HL2 (4) under aerobic conditions with iron(II) acetate. In addition, 5 can be converted to complexes 6-8 by co-ligand exchange or nickel(II) acetate (Fig. 6) [7,8]. The asymmetrically substituted µ3-oxo centered complexes [MIIFeIII2O(L2)3(O2C-R)3] (MII=FeII, NiII; R=Me, Ph) (5-8) possess a metal-to-ligand-to-co-ligand ratio (M:L:co-L) of 1:1:1, and the absence of a counterion requires intramolecular charge compensation, i.e. mixed-valence character for 5-8. Fig. 6
Stereoview of 6 with numbering of the iron ions: FeII blue, FeIII gold, C white, N blue (small), O red, S yellow Fig. 7 The X-ray structure of [FeIIFeIII2O(L2)3(OBz)3] (6) reveals: The iron centers Fe1/Fe2 are linked by two ligands (L2)-, Fe2/Fe3 by one (L2)- ligand and a benzoate ion, and Fe1/Fe3 by two benzoate bridges. As a consequence, all three iron ions in the mixed-valent complex 6 are differently octahedrally coordinated (Fig. 7). The effect of the (OAc)- co-ligands on the {FeIIFeIII2O} core in 5 is different from the effect of the (OBz)- co-ligands on the iron core in 6, leading to the situation that in 6 the difference in coordination among iron sites is larger than in 5. This situation is reflected in the Mössbauer spectra (Fig. 8).
The Mössbauer spectra of [FeIIFeIII2O(L2)3(O2C-R)3] (5,6)(R=Me, Ph) are shown in Fig. 8 [7,8]. The spectra of 5 and 6 at 300 K are almost identical; both exhibit two quadrupolar doublets with an area ratio of 1:2 for the FeII and the FeIII ions. However, at 4.2 K the Mössbauer spectra of 5 and 6 differ considerably. Unlike for 5 the spectrum of 6 shows at 4.2 K a rather broad signature of the FeII doublet as compared with the FeIII doublet, i.e. the experimental data (inset in Fig. 8d) are best fitted by three doublets (DEQ=2.77, 2.66, 2.66 mms-1, d=1.19, 1.12, 0.96 mms-1, G=0.25, 0.25, 0.25mms-1), which represent FeII with a relative area ratio of 11,11,11 %, respectively (Table 1) [7]. The presence of partial electron (valence) delocalisation at 4.2 K in the present case is excluded, since the hetero-nuclear all-ferric complexes [NiIIFeIII2O(L2)3(O2C-R)3] (7,8) (R=Me, Ph) exhibit comparable isomer shifts at 77 K, i.e. 0.50 mms-1 and 0.49 mms-1, as the ferric sites in 5 and 6 at 4.2 K (Table 1). Fig. 8
As a result of the different coordination of the iron sites (Fe1)3N/3O, (Fe2)4N/2O, (Fe3)2N/4O in [FeIIFeIII2O(L2)3(OBz)3] (6), there exist evidently three configurations with unlike energies (Fe1)II(Fe2)III(Fe3)III, (Fe1)III(Fe2)II(Fe3)III, (Fe1)III(Fe2)III(Fe3)II for 5 and 6. Even if the difference among these energies is small and can be overcome at elevated temperature by thermal activation, at low temperature one expects that only the configuration with the lowest energy is populated, corresponding to only one FeII doublet (instead of three doublets) in the Mössbauer spectrum at 4.2 K. However, assuming a slow electron-tunneling exchange mechanism in 6, one might arrive at a situation, at which all three configurations remain nearly equally populated, even at very low temperature. Thermal fluctuations at 300 K then equalise the ligand-field strength around each iron site in 5 and in 6, such that it is not possible to resolve the different (Fe1)II, (Fe2)II, (Fe3)II components in the Mössbauer spectra.
Summary Mixed-valent oxo-centered trinuclear metal complexes provide model systems for systematic studies of the phenomenon of electron (valence) delocalisation. Such investigations have shown that the rate of electron transfer is dramatically affected by the environment of the complex, especially by solvent and/or packing, and by the nature of the ligands directly coordinated to the metal sites. References [1] R.W. Saalfrank, S. Trummer, H. Krautscheid, V. Schünemann, A.X. Trautwein, S. Hien, C. Stadler and J. Daub, Angew. Chemie. Int. Ed. Engl. 35, 2206 (1996) [2] D.N. Hendrickson, in Mixed Valency Systems: Applications in Chemistry, Physics and Biology, K. Prassides (Ed.), Kluwer, Dordrecht, 1991, p. 67 [3] C.-C. Wu, H.G. Jang, A.L. Rheingold, P. Gütlich and D.N. Hendrickson, Inorg. Chem. 35, 4137 (1996) [4] V. Coropceanu, V. Schünemann, C. Ober, M. Gerdan, A.X.Trautwein, J. Köhler and R.W. Saalfrank, Inorg. Chim. Acta 300-302, 875 (2000) [5] D.P.E. Dickson and F.J. Berry, Mössbauer Spectroscopy, Cambridge University Press, Cambridge, UK, 1986, p. 88 [6] C. Stadler, J. Daub, J. Köhler, R.W. Saalfrank, V. Coropceanu, V. Schünemann, C. Ober, A.X. Trautwein, S.F. Parker, M. Poyraz, T. Inomata and R.D. Cannon, J. Chem. Soc., Dalton Trans. 3373 (2001) [7] R.W.Saalfrank, A. Scheurer, U. Reimann, F. Hampel, C. Trieflinger, M. Büschel, J. Daub, A.X. Trautwein, V. Schünemann and V. Coropceanu, Chem. Eur. J. 11, 5843 (2005) [8] R.W. Saalfrank, A. Scheurer, K. Pokorny, H. Maid, U. Reimann, F. Hampel, F. W. Heinemann, V Schünemann and A.X. Trautwein, Eur. J. Inorg. Chem. 1383 (2005)

















