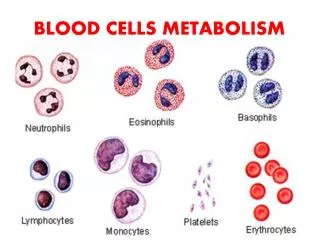
Download

1 / 65
730 likes | 1.03k Views
RED. Blood cells. HAEMOGLOBINOPATHIES. هموگلوبین هموگلوبین مهمترین وفراوانترین پروتئین موجـود در گلبولهای قرمز بوده و نقش اساسی در حمل ونقل O2 ،CO2 و پروتون دارد. میوگلوبین نیز پروتئینی مشابه هموگلوبین بوده که درون سلولهای عضلانی قرار گرفته و در انتقال O2 دخالت دارد. ساختمان هموگلوبین

E N D
RED Blood cells HAEMOGLOBINOPATHIES
هموگلوبین هموگلوبین مهمترین وفراوانترین پروتئین موجـود در گلبولهای قرمز بوده و نقش اساسی در حمل ونقل O2 ،CO2 و پروتون دارد. میوگلوبین نیز پروتئینی مشابه هموگلوبین بوده که درون سلولهای عضلانی قرار گرفته و در انتقالO2 دخالت دارد. ساختمان هموگلوبین مولکول هموگلوبین شامل دو بخش میباشد: 1- بخش پروتئینی موسوم به گلوبین که دارای چهار زنجیره پلی پپتیدی می باشد (تترامر است) 2- بخش غیر پروتئینی موسوم به هِم(heme) که به آن گروه پروستتیک مولکول هموگلوبین می گویند. هِم عبارتست از یک حلقه پورفیرینی که یک اتم آهن دو ظرفیتی در مرکز آن قرار گرفته است.هر مولکول هموگلوبین دارای یک مولکول گلوبین و4 مولکول هِم می باشد، بطوریکه هر مولکول هِم به یکی از زنجیره هـــای گلوبین متصل میگردد. هر مولکول هِم قادر است یک مولکولO2 را جابجا کند، لذا هر مولکول هموگلوبین توانایی حمل چهار مولکول O2 را داراست.در مولکول میوگلوبین دو بخش گلوبین و هِم مشاهده می شود، اما مولکول گلوبین آن از یک زنجیره پلی پپتیدی ساخته شده است که به یک مولکول هِم متصل می گردد، لذا مولکول میـــوگلوبین تنها توانایی انتقال یک مولکولO2 را دارد.
مولکول هِم مسئول رنگ قرمز هموگلوبین و میوگلوبین است و همانطوریکه گفته شد یک اتم آهن دو ظرفیتی در مرکز آن قرار گرفته است. اگر اتم آهن دو ظرفیتی(Fe²+) به آهن سه ظرفیتی(Fe³+) اکسید شود، عملکرد مولکول از بین خواهد رفت. اتمهای آهن فرو Fe++با شش پیوند با مولکول ارتباط دارند : 4 پیوند با نیتروژنهای پیرول هم 1 پیوند با نیتروژن ایمیدازول مربوط به هیستیدین زنجیره گلوبین 1 پیوند بطور برگشت پذیر با اکسیژن
سنتز هموگلوبیننحوه سنتز هموگلوبین شامل دو مرحله سنتز گلوبین و سنتز هِم می باشد. سنتز گلوبین در سیتوپلاسم و روی ریبوزومها صورت مــی پذیرد و سنتز هِم داخل میتوکندری انجام می گیرد.سنتز گلوبین: سنتز زنجیره هــای گلوبین که دارای ساختمان پلی پپتیدی می باشند، نیازمند ژن است. زنجیره پلی پپتیدی مولکول گلوبین شامل انواع α ،β ،γ،δ، ζ و ε بوده که هر کدام دارای ژنهای خاص خود می باشد که روی کروموزوم بخصوصی قرار گرفته است:زنجیره β ، γ ، δ روی کروموزوم 11 با 146 اسید آمینه در صورتیکه ژن زنجیره α با 141 اسید آمینه روی کروموزوم 16 قرار دارد.لازم به ذکر است که:1. محل قرار گیری ژنهایβ وδ روی کروموزوم11 نزدیک هم است.2. ژنهایε وζ فقط در دوران جنینی و در کیسه زرده فعالند.3. منظور از دو یا یک عدد ژن روی کروموزوم، قرار گرفتن روی یک رشته از یک جفت کروموزوم است.4. احتمال موتاسیون در زنجیرهβ بیش از زنجیرهα است، زیرا ژنهای آن فقط یک عدد است.5. در هموگلوبینهای طبیعی مولکول گلوبین شامل دو جفت زنجیره می باشد، مثلاً α2β2
نحوه انتقالO2 توسط مولکول هموگلوبینبه هنگام تنفس O2 هوای حبابچـه های ریوی از طریق انتشار (دیفیوژن) جذب خون مویرگی ریه ها می شود، که بیش از97 درصد اکسیژن گیری خون توسط هموگلوبین انجام می شود. هوایی که ریه ها را ترک کرده و وارد سیستمهای سرخرگی می شود دارای فشار سهمی95میلیمتر جیوه است. فشارO2 در مویرگهای سیستمیک و مایعات بین سلولی به طور متوسط mmHg 40 است که عملاً در خون وریدی نیز با همین فشار تا ریه ها حمل می شود، تا اینکه توسط تهویه آلوئولی ریه مجدداً فشار آن از 45 به 95 برسد. مولکول هموگلوبین بعلت ساختمان فضایی ویژه وتخصص یافته اش، صفات جالبی را در مورد تمایل بهO2 در فشارهای مختلف از خود نشان می دهد. این صفات را می توان به کمک منحنی تغییرات اشباع هموگلوبین با تغییرات فشارO2 بررسی کرد.حتی اگر فشار اکسیژن حبــابچه های ریوی بینmmHg60 و بالاتر تغییر کند، بیش از90% هموگلوبین با اکسیژن اشباع خواهد شد. از طرف دیگر در بافتها یعنی در محدودهmmHg40 حتی کوچکترین افت فشار O2 در مایعات بین بافتی، منجر به کاهش شدید اشباع شدگی هموگلوبین با O2 خواهد شد، به عبارتی دیگر کاهش مختصری در فشارO2 در بافتها موجب خواهد شد که Hbحجم زیادی از O2 را آزاد کرده و کمبود اکسیژن بافتی را جبران کند.
منحنی تغییر اشباع Hbبا O2 تحت تأثیر برخی عوامل ممکن است به سمت راست یا چپ منحرف شود. به هنگام انحراف به راست تمایلHbبرای آزادسازی O2 بیشتر است و به هنگام انحراف به چپ تمایل آن برای آزاد کردن O2 کمتر است. مهمترین عامل منحرف کننده منحنی به سمت راست عبارتست از کاهش pH (اسیدی شدن محیط). این خاصیت باعث خواهد شد که اگر pH بافتی بعلت افزایش متابولیسم کاهش یابد (مثلاً فعالیت عضلانی سنگین و تولید اسید لاکتیک)، آزاد شدن O2 در مجاورت بافت ها نیز بیشتر می شود، که این امر به نوبه خود جهت رفع نیازهای متابولیکی ضروری می باشد. افزایش آزاد شدن O2 به هنگام کاهش pH محیط را اثر بوهر می نامند.عوامل دیگری که می توانند منحنی اشباع را به سمت راست منحرف نمایند عبارتند از: افزایش فشار CO2 ، افزایش دما و افزایش غلظت 2و3- دی فسفو گلیسرات (2-3-DPG ماده ای است که از فرآورده های متابولیسم گلیکولیز می باشد که در شرایط بیهوازی تشکیل می گردد).عواملی که منحنی را به سمت چپ منحرف می کنند: افزایشpH (قلیایی شدن محیط)، کاهش دما و کاهش غلظت 2-3-DPG.هنگامیکه هموگلوبین اولینO2 را کسب کرد، دومین و سومین O2 را به سادگی کسب میکند. یعنی زمانیکه هموگلوبین اولین O2 را کسب می کند، این پدیده در هِم های مجاور آن هِمی که اولین O2 را گرفته بود اثر گذاشته و هِمهای مجاور با سهولت بیشتر و اختلاف فشار کمتری O2 خود را بدست می آورند. برعکس اگر هموگلوبین4 عدد O2 داشته باشد واولین O2 را از دست بدهد، این مسئله بر هِم های مجاور اثر گذاشته و در نتیجه آنها راحت تر O2 را از دست می دهند. به این پدیده Heme-HemeIntractionیاAllostric Effect یا اثر تعاونیCooperative Effect) )می گویند.
هموگلوبینوپاتی 2گروه اصلی از اختلالات ارثی هموگلوبین وجود دارد: 1-واریان های ساختاری همراه با نقص هایی در ساختار هموگلوبین 2-تالاسمی ها همراه با نقص هایی در سنتز یک زنجیره گلوبینی انواع هموگلوبینهای نرمالهموگلوبینهای نرمال و طبیعی را می توان به دو گروه تقسیم نمود:الف) هموگلوبینهای پیش از تولد شامل:هموگلوبینهای رویانیEmberyonic Phase))، سه ماه اول جنینی، شامل:Hb Portland با فرمول ζ2γ2Hb Gower I با فرمول ζ2ε2Hb Gower II با فرمول α2ε2هموگلوبینهای جنینی، بعد از سه ماه اول، شامل:Hb F با فرمولα2γ2Hb F1 با فرمول α2γ2 N-Acetyleب) هموگلوبینهای پس از تولد و دوران بلوغ، شامل:Hb A با فرمول α2β2Hb A2 با فرمول α2δ2Hb F با فرمول α2γ2
در افراد بالغ حدود 98-95% راHb A1 ، حدود 5-3% را Hb A2 وHb F تشکیل می دهد (5/2%=A2 و F حدود1 درصد). :Hb A هموگلوبین اصلی طبیعی در بزرگسالان با فرمول ساختمانα2ß2و مقدار طبیعی آن در افراد نرمال 92% می باشد .بیضی شکل است و همراه 4گروه هم در سطح ملکول،در ترکیب برگشت پذیر با اکسیژن عمل می کند. Hb F :هموگلوبین اصلی جنین و شیر خوار نوزاد است و فرمول ساختمانی آنα2γ2می باشد و مقدار طبیعی آن در افراد نرمال 1>% می باشد . موارد افزایش: حاملگی طبیعی – اختلالات اریتروپوئیز همچون نارسایی مزمن مغز استخوان هموگلوبین F با بار منفی را Hb F1 می گویند (استیله شدن انتهای آمینو اسید زنجیره γ) Hb F همچنین دارای دو نوع زنجیره گاما می باشد که در یک اسید آمینه در موقعیت 136 اختلاف دارند (آلانین یا گلایسین ) یک پلی مورفیسم شایع زنجیره γوجود دارد که در ان تیروزین در موقعیت 75 جایگزین ایزولوسین می شود.این واریان Hb F ساردینیا(Sardinia) نامیده میشود و بدون ناهنجاری عملکردی است.
در طول زندگی جنینی تمام گویچه های قرمز Hb F را تولید نموده و حاوی ان هستند در صورتی که در بزرگسالان تنها 3-5% گویچه های قرمز چنین حالتی دارند.این گویچه های قرمز،سلول های F نامیده میشوند.بالاترین سطح Hb F در لوسمی میلومونوسیتی نوجوانان(JMML)،کم خونی فانکونی و اریترولوکمی(30-50%) یافت میشوند. Hb A2 :این هموگلوبین 1/5 تا 3/5 درصد هموگلوبین طبیعی محسوب می شود .وفرمول ساختمانی آن α2δ2 می باشد .دو زنجیره دلتا آن از زنجیره های بتا فقط در 8 اسید آمینه از 146 اسید آمینه اختلاف دارند . موارد افزایش : بتا تالاسمی – هیپرتیروئیدیسم – کم خونی مگالو بلاستیک – سیکل سل آنمی – واریانت های هموگلوبین ناپایدار موارد نرمال یا کاهش یافته : بتا دلتا تالاسمی – آلفا تالاسمی تریت موارد کاهش : بیماری هموگلوبین اچ – فقر آهن – آنمی سیدروبلاستیک –آنمی آپلاستیک و . . . . هموگلوبینهای دوره رویانی شامل : Gower │ با فرمول ساختمانی ζ2ε2 Gower ││ با فرمول ساختمانی α2ε2 Portland با فرمول ساختمانیζ2γ2 این هموگلوبین ها در رویان و جنین طبیعی انسان با دوره بارداری کمتر از 3 ماهگی یافت می شوند .
هموگلوبينهای گليکوزيله : 5% از HbA در نتيجه الحاق قندها به باقيمانده های سرين، آسپاراژين و هيدروکسی ليزين دستخوش گليکوزيلاسيون پس از ترجمه می شوند . هموگلوبين های گليکوزيله تحت عنوان: HbA1a کمتر از 1% HbA1b کمتر از 2% HbA1c حدود 3% (اين هموگلوبين حاوی يک ملکول گلوکز است و در بيماران مبتلا به ديابت قندی 2 تا 3 برابر افزايش مييابد .واز آن به صورت شاخصی از کنترل متابوليک ديابت در طی دو تا سه ماهه گذشته استفاده می شود همچنين ممکن است بطور کاذب در شرايط همراه با گردش سريع گلبول قرمز مثل کم خونی همولتيک کاهش يابد .)
واریان های ساختاری هموگلوبین تا سال 2005، 893 گونه ساختاری Hbشناسایی شده که بیش از 90% آنها بعلت اختلاف اسید آمینه ای واحد در یکی از زنجیره های پلی پپتیدی α یا β هستند .در 10%باقی مانده زنجیره پلی پپتیدی در نتیجه خطاهای خاطمه،جهش های تغییر چارچوب،الحاق ها، حذف ها و یا زنجیره های هیبرید یا جوش خورده،بصورت غیر طبیعی کوتاه یا بلند می باشد. براي نام گذاري هموگلوبينوپاتي ها، نوع اسيد آمينه جايگزين شده و شماره آنرا در سمت راست زنجيره و گلوبين مي نويسند مثلاً در مورد Hbs a2b26Val يعني در هموگلوبين S،اسيد آمينه والين به جاي اسيد آمينه اصلي در موقعيت 6 زنجيزه βگلوبين قرار گرفته است اسيد آمينه اصلي در اين فرمول ذكر نمي شود اما در مورد Hbsو نيز Hbcگلوتاميك اسيد استهموگلوبين هاي E,D,C,S معروف ترين (بتا) هموگلوبينوپاتي ها مي باشند.هموگلوبينوپاتي ها معمولاً از نوع β هستند و انواع با درگيري زنجيره آلفا و گاما كم ياب هستند و علائم باليني خاص نيز حتي در حالت هموزيگوت ندارند. هموگلوبين هاي غير طبيعي بجز موارد اكتسابي مانند سولف هموگلوبين و مت هموگلوبين، به طور ارثي بروز مي كنند.
در حالت هموزيگوت يك فرد هر دو ژن غير طبيعي را براي توليد هموگلوبين غيرطبيعي دارد يعني يكي از ژن هاي معيوب را از پدر و ديگري را از مادر به ارث برده است .در حالت هتروزيگوت فرد حامل يك ژن سالم و يك ژن معيوب است .به حالت هتروزيگوت(trait)خصيصه مي گويند و به حالت هموزيگوت(بيماري) گفته مي شود.به حالت هتروزيگوت به آن علت بيماري نمي گويند كه در اين وضعيت علائم باليني خاص وجود ندارد و ناهنجاري خوني نيز حداقل است .
سندروم های هموگلوبین غیر طبیعی در واریان های ساختاری زنجیره بتای هموزیگوت،هر 2 الل زنجیرهβ غیر طبیعی،وجود دارند.لذا هیچ زنجیره β طبیعی(در نتیجه هیچ Hb A)تولید نمی شود.از انجایی که ژن های α و γوδ طبیعی هستند Hb F و Hb A2 تشکلیل شده و ساختاری طبیعی دارند و مقدار ان ها ممکن است افزایش یافته باشد. در واریان های ساختاری زنجیره بتای هتروزیگوت، هموگلوبین غیر طبیعی علاوه بر هموگلوبینA و F و A2وجود دارد.سطح هموگلوبین غیر طبیعی به سرعت سنتز و پایداری ان و همچنین به اشتیاق ان برای تشکیل دادن دایمر با زنجیره الفا بستگی دارد.
اختلالات داسی شونده خصیصه سلول داسی Hb AS وضعيت هتروزيگوت HBS شايع ترين هموگلوبينوپاتي در ايالات متحده است.اين وضعيت در حدود 8%امريكايي هاي افريقايي تبار و حدود 45%از مردمان نواحي خاصي از غرب افريقا وجود دارد.اين اين بيماري در سرتاسر ناحيه زير صحراي افريقا و با شيوع كم تر در نواحي مديترانه اي خاورميانه و هند يافت ميشود. خصيصه HBS يك حالت كاملا خوش خيم و بدون علايم باليني يا ناهنجاري هاي خون شناختي است.در اكثر موارد خطر جزئي است و در شرايط خاصي ديده ميشود.فشار هاي شديدا پايين اكسيژن مي تواند باعث نشانه گيري فرايند داسي شدن شود و تعداد كمي از افراد در زماني كه در ارتفاعات بسيار بالا و در هواپيما هاي بدون تنظيم فشار هوا پرواز كرده اند،دچار انفاركتوس طحالي شده اند.اندكي افزايش در بروز هماچوري،نقص در توانايي تغليظ ادرار،و باكتري اوري در خانم ها گزارش شده است.خصيصه سلول داسي به بچه ها در برابر اثرات كشنده مالارياي فالسيپاروم حالت محافظتي مي بخشد كه توجيهي براي توزيع عمده HBS در افريقاي جنوبي است.
گسترش خوني بجز چند سلول هدف طبيعي بنظر ميرسد.شمارش سلول هاي خوني طبيعي و ازمايش داسي شدن گويچه ها مثبت است و تقريبا تمام گويچه هاي قرمز خون بطور يكدست داسي ميشوند.ازمايش انحلال پذيري مثبت است. گستره خون محیطی خصیصه داسی شکل.تعدادی سلول هدف و میکروسیتوزیس خفیف مشاهده میشود.
جداسازي هموگلوبين،60% هموگلوبين A،40% HbS، HbFطبيعي و HbA2 طبيعي يا اندكي افزايش يافته تا 5/4% را نشان ميدهد.نسبت HbS در حضور تالاسمي a كاهش مي يابد:هنگامي كه يك ژن a حذف شود، HbS كم تر از 35% و هنگامي كه 2ژن a حذف شود كم تر از 29% خواهد بود.سلول ها ميكروسيت و هيپوكروم هستند. HbS در فقر اهن و فولات نيز ممكن است كاهش يابد.بطور باليني تركيب خصيصه داسي شكل و خصيصه aتالاسمي خوش خيم بوده و احتمالا با كم خوني ميكروسيتيك خفيف به علت خصيصه تالاسمي مي باشد. بيماري سلول داسي شكل(Hb SS) بيماري HbS هموزيگوت يك كم خوني هموليتيك مزمن جدي است كه ابتدا در اوايل دوره كودكي تظاهر مي كند و اغلب قيب از 30 سالگي كشنده است.در هموگلوبين S اسيد گلوتاميك در ششمين جايگاه بر روي زنجيره β با والين جايگزين مي شود(β6glu→val).هموگلوبين S در هنگام اكسيژن گيري كامل،ازادانه محلول است.هنگامي كه اكسيژن از HbS برداشته مي شود، HbS پلي مريزه شده و اجسام تاكتوئيدي(كريستال هاي مايع) را تشكيل مي دهد كه سخت است و سلول را بشكلي در مي اورد كه به ان اطلاق مي شود(داسي شكل).
عوارض در اوايل كودكي تورم دردناك و دوطرفه پشت دست ها و پاها بخاطر داسي شدن گويچه ها و انسداد مويرگي رخ مي دهند كه تحت عنوان سندروم دست-پا يا التهاب انگشتان سلول داسي شناخته مي شود.
طحال مركز 3 عارضه است:بحران احتباس كه به انبار شدن ناگهاني خون و بزرگ شدن سريع طحال اشاره دارد و منجر به شوك كم حجمي خون مي شود.بي عملكردي طحال شامل پاسخ ناكافي به انتي بادي تحت بعضي شرايط و اختلال در توانايي سيستم رتيكواندوتليال در پاكسازي باكتري ها و مواد ذره اي ازخون.اين موضوع مي تواند تا حدي عفونت هاي سالمونلايي و پنوموكوكي را كه در كودكان مبتلا به كم خوني سلول داسي شايع است،توجيه كند.رخدادهاي انسداد رگي باعث انفاركتوس پيشرونده،فيبروز و چروكيدگي طحال يا اصطلاحا اتواسپلنكتومي مي شوند. Splenomegaly
بحران هاي انسداد عروقي حملات ناتوان كننده اي از دردشكمي،استخواني و مفصلي همراه با تب مي باشند،كه احتمالا به خاطر گيركردن توده هايي از سلول هاي داسي در رگ هاي خوني كوچك رخ مي دهد. تشخيص:كم خوني نرموكروميك و نرموسيتيك است،پلي كرومازي افزايش مي يابد و گويچه هاي قرمز هسته دار وجود دارند.سلول هاي داسي هميشه در گسترش يافت مي شوند.سلول هاي هدف فراوان هستند،اجسام هاول ژولي و پاين هايمر در نتيجه عدم وجود طحال در كودكان بزرگ تر و بزرگسلان به طور هميشگي ديده مي شود.
چنانچه اخيرا به بيمار خون تزريق نشده باشد،هيچ هموگلوبين A يافت نمي شود.بيش از 80%HbS،1-20%HbF،2-5/4% HbA2 وجود دارد. HbS و چنديدن هموگلوبين DوG درPHقليايي حركت الكتروفورتيك مشابه دارند،اما تنها HbS ازمايش داسي شدن مثبت بدست مي دهد. بيماري هموگلوبين SC شيوع ان در امريكايي هاي افريقايي تبار تقريبا با بيماري HbSS يكسان است.منجر به يك كم خوني هموليتيك خفيف مشود.بحران ها در اينجا وفور كم تري دارند و كم تر دردناك هستند.شيوع ان در دوره كودكي است اما ممكن است تا سنين بالاتر زندگي هم كشف نشود.برخلاف ظاهر رنجور در بيماري داسي شكل هيكل فرد در اينجا طبيعي يا تنومند است.اسپلنومگالي ممكن است تنها يافته معاينه فيزيكي باشد.خستگي، تنگي در نفس در حين فعاليت،عفونت هاي مكرر دستگاه تنفسي فوقانيفحملات يرقان خفيف و درد مفاصل ديده ميشود.درد مداوم مفصل ران و كمر بهمراه نكروز اسپتيك سر استخوان ران ممكن است وجود داشته باشد.هماچوري ناشي از انفاركتوس مدولاري كليه و انفاركت هاي طحالي توصيف شده اند.در حاملگي بحران ها فراوان ترند و تمايل افزايش يافته اي براي ترومبوز وجود دارد كه ميتواند باعث ترومبوامبوليسم وسيع و مرگ ناگهاني بدنبال زايمان شود.ميزان بروز بالاتري از بيماري هاي شبكيه(رتينوپاتي) نسبت به SS وجود دارد.
كم خوني از متوسط تا بسيار خفيف متغير است و نرموكروميك تا نرموسيتيك ميباشد.انيزوسيتوز و پوئي كيلوسيتوز خفيف تا شديد ميباشد.تعداد سلول هاي هدف بسيار زياد و تا 85%اريتروسيت ها شامل ميشود.گويچه هاي داسي شكل خپل و زاويه دار در گسترش خوني وجود دارند.ازمايش داسي شدن مثبت است،HbS،50%هموگلوبين تام است و HbC اندكي كم تر است.
HbS / تالاسمي بتا اين بيماري پس از HbSS و HbSC از نظر شيوع سومين اختلال داسي شونده در امريكايي هاي افريقايي تبار است و شايع ترين اختلال داسي شونده در مردمان مديترانه اي است. در تالاسمي HbS / B0،هموگلوبين A(HbA) وجود ندارد، HbS75تا90%، HbF 5تا20%و HbA2 4تا6%است.از نظ باليني و خون شناسي ،مشابه بيماري سلول داسي شكل است.بجز در مورد طحال كه پس از دوران كودكي و در طي زندگي بزرگسالي طحال بزرگ باقي مي ماند،تفاوت اصلي اين است كه در تالاسمي B0/ HbS،MCVوMCH كاهش مي يابد و HbA2 ممكن است افزايش چشمگيري داشته باشد.براي افتراق واضح اين دو بيماري از هم اغلب بررسي خانوادگي ضرورت دارد.در گسترش خوني ميكروسيتوز قابل توجه،هيپوكرومي متغير و تعداد زيادي سلول هدف وجود دارد.به عنوان پيامدي از كاهش هموگلوبين سلولي،شكل گيري پلي مرف هاي HbS با سرعت اهسته تري انجام ميشود و تعداد كم تري سلول داسي شكل در مقايسه با HbSS در گسترش خوني وجود دارد.
در تالاسمي B+/ HbS،هموگلوبين A15-30% است. HbS بيش از 50% و HbF 1-20% و HbA2 4-6% است.اگرچه از نظر باليني اين افراد ممكن است شبيه بيماران مبتلا به خصيصه سلول داسي شكل(HbAS) باشند،اما در تالاسمي B+/ HbS،مقدار HbS هميشه بيش تر از HbA است درحالي كه در HbAS،مقدار HbA هميشه بيش تر از HbS است. HbSS / تالاسمي a 30-40% از بيماران مبتلا به بيماري كم خوني سلول داسي شكل براي تالاسمي a+ هتروزيگوت و 2-3% هموزيگوت هستند.MCVوMCH كاهش مي يابد.در مقايسه با كم خوني سلول داسي شكل،سطح هموگلوبين بالاتر و شمارش رتيكولوسيت پايين تر است.در گسترش خوني شبيه به B0/ HbS، سلول هاي داسي ناشايع هستند.
بيماري هموگلوبين SD Hb S/D-Los Angeles بيماري SD شبيه به كم خوني سلول داسي شكل تظاهر ميكند اما شدت كم تري دارد و لذا ممكن است شبيه به بيماري SC نيز باشد.در الكتروفورز قليايي متداول،الگوي الكتروفورز اين بيماري غير قابل افتراق از كم خوني داسي است ، زيرا Hb S و Hb D را نمي توان از يكديگر جدا كرد.الكتروفورز ژل اگار در PH=6/2 ، Hb S و Hb D را جدا خواهد نمود. O Arab/Hb S افراد هتروزيگوت مركب Hb S و HbOArab(β121glu→lys)، يك اختلال داسي شونده شديد دارند. HbOArab در مردم سياه پوست و عرب يافت ميشود. HbE و تعداد كمي از ديگر واريان هاي β با شيوع كم تر نيز در همراهي با HbS باعث اختلالات داسي شونده مي شوند.همزماني يك واريان زنجيره a و خصيصه سلول داسي شكل منجر به اختلال داسي شونده نمي شوند.
ديگر واريان هاي شايع زنجيره β خصيصه Hb C هموگلوبين C((β6glu→lys در افريقاي غربي شايع است و در حدود 2-3%سياه پوستان وجود دارد.وضعيت هتروزيگوت(HbAC) بدون نشانه و بدون كم خوني و همراه با MCVطبيعي يا اندكي كاهش يافته و طول عمر طبيعي گويچه قرمز مي باشد.ممكن است MCHC اندكي افزايش يابد.سلول هاي هدف در لام خوني وجود دارد.HbC حدود 40%هموگلوبين تام را تشكيل ميدهد.هرگاه ميكروسيتوز قابل توجهي وجود داشته باشد معمولا بخاطر همزماني تالاسمي a ميباشد و HbC به 38%،32%،24% به ترتيب در بيماران با 1،2،3 ژن الفا كاهش مي يابد.
بيماري هموگلوبين C بيماري هموگلوبين C هموزيگوت يك كم خوني هموليتيك خفيف همراه با اسپلنومگالي مي باشد كه اغلب بدون علايم است اما گاهي اوقات منجر به يرقان و ناراحتي شكمي مي گردد.در امريكا 2%سياه پوستان اين بيماري را دارند.رتيكولوسيتوز با توجه به ميزان كم خوني پايين است.كم خوني عمدتا پيامدي از ميل پيوندي پايين Hb C براي اكسيژن است و نبايد مداوا شود.MCV طبيعي يا كاهش يافته و MCHC طبيعي يا افزايش يافته است.تعداد زيادي سلول هدف همراه با مخلوطي از ميكرواسفروسيت ها و پلي كرومازي جزئي در خون مشاهده ميشود.براي تشخيص بلورهاي شش گوشه يا ميله اي شكل ممكن است در گسترش رنگ شده بخصوص پس از طحال برداري يا پس از خشك شدن اهسته گسترش در اريتروسيت ها ديده شوند.برخلاف HbS،كريستال هاي HbC در PO2(فشار نسبي اكسيژن)پايين تمايل به حل شدن دارند و باعث بيماري انسداد عروقي نمي شوند. HbFبه 2-4% افزايش مي يابد و HbA2ممكن است بطور خفيفي افزايش يابد،مابقي هموگلوبين،HbC است و HbA وجود ندارد.
تالاسمي B+/ HbC عمدتا در سياه پوستان روي مي دهد و بيماري به استثنا كم خوني در طي حاملگي گرايش به ايجاد ناتواني هاي اندكي دارد.گويچه هاي قرمز انديس هاي ويژه خصيصه تالاسمي B را دارند اما در اينجا انيزوسيتوز وجود دارد و سلول هدف 20-50% هستند.65-80% HbC،16-30% HbA و 2-5% HbF وجود دارد. تالاسمي B0/ HbC افراد با اصل و نسب مديترانه اي با ژنوتيپ B0 يا B+شديد معمولا داراي يك كم خوني هموليتيك نسبتا شديد هستند.افتراق تالاسمي B0/ HbC از بيماري HbC ممكن است مشكل باشد زيرا در هر دوي اين بيماري ها HbA وجود ندارد و درمورد سطوح HbF و HbA2 همپوشاني وجود دارد. HbA2 حدودا 2برابر مقدار طبيعي و HbF به 3-10%افزايش مي يابد.
HbE((β26glu→lys اين هموگلوبين احتمالا شايع ترين هموگلوبين غير طبيعي در جهان است و پس از هموگلوبين S وc شايع ترين هموگلوبين غير طبيعي در ايالات متحده است.در اسيا نيز در افراد با اصل و نسب تايلندي و برمه اي يافت ميشود در سفيدپوستان و سياه پوستان نيز وجود دارد. HbE با فنوتيپ تالاسمي بتا و همچنين يك زنجيره گلوبيني غير طبيعي از نطر ساختاري ارتباط دارد.جهشي كه منجربه جايگزيني اسيد امينه مي شود همچنين يك محل اتصال اشكار(Cryptic splice site) را نيز فعال مي كند كه با محل اتصال در RNAطبيعي رقابت مي كند و در نتيجه RNAي پردازش يافته طبيعي،كاهش مي يابد. HbE در ازمايش تقليب حرارتي و با ايزوپروپانول بطور غير طبيعي رسوب مي كند، با اين وجود اين موضوع هنوز هيچ اهميتي در داخل بدن ندارد،طول عمر گويچه هاي قرمز در بيماري HbE طبيعي است.