Download

1 / 26
320 likes | 662 Views
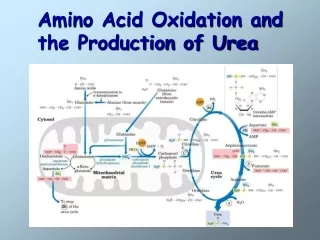
Amino acid oxidation and the production of urea. Catabolism of proteins and aa nitrogen. How the nitrogen of aa is converted to urea and the rare disorders that accompany defects in urea biosynthesis Normal condition- nitrogen intake match nitrogen excreted

E N D
Catabolism of proteins and aa nitrogen • How the nitrogen of aa is converted to urea and the rare disorders that accompany defects in urea biosynthesis • Normal condition- nitrogen intake match nitrogen excreted • Positive nitrogen balance- an excess of ingested over excreted nitrogen- during growth and pregnancy • Negative nitrogen balance – output exceeds intake- during surgery, advanced cancer or malnutrition
Oxidative degradation occur in 3 diff metabolic circumstances: • During normal synth and degradation of cellular protein- aa that are released from protein breakdown are not needed for new prot synthesis-degraded • Taking a protein rich diet- aa intake exceeds the body’s need for prot- degraded • During starvation or in uncontrolled DM – when carb cannot be utilized, proteins are used as fuel
Under all this metabolic conditions- aa lose their amino groups to form α-ketoacids (the carbon skeleton of amino acids) • The α-ketoacids undergo oxidation to CO2 and H20 and provide 3-4C for gluconeogenesis
Metabolic fates of amino groups • Metabolic fates carbon skeletons
Metabolic fates of amino groups • Degradation of ingested proteins to aa occurs in gastrointestinal tract • Entry of protein dietary will stimulate the hormone gastrin – will stimulate the production of HCl- kill most bacteria, denaturing agent for protein, unfolding globular proteins, and make internal peptide bonds more accessible to enzymatic hydrolysis • Pepsin is activated to cleave the polypeptide to smaller peptide
Dietary protein is enzymatically degraded to amino acids • Once protein are degraded to amino acids, aa are transported to liver- to remove the amino groups – by aminotransferases or transaminases • The amino groups are transferred α-ketoglutarate- forming glutamate
Glutamate releases its amino group as ammonia in the liver • Amino groups must be removed from glutamate for excretion • In hepatocytes, glutamate is transported from cytosol to mitoch – undergoes oxidative deamination- cat by glutamate dehydrogenase produced NH4+ • NH4+ is transported by glutamine to liver to be secreted thru urea cycle
Excretion of excess nitrogen • Excess nitrogen – excreted in one of three forms: ammonia (as ammonium ion), urea and uric acid • Fish – excrete ammonia – protected by the toxic activity through excretion and rapid dilution by environment • Terrestrial animals- excrete urea- water soluble compounds • Birds- excrete uric acid – insoluble in water- to avoid excess weight • High blood urea level as a consequence (not a cause) of impaired renal fx
Urea cycle • Central pathway in nitrogen metabolism- urea cycle • Start with the reaction of ammonium ion and C02 to produce carbamoyl phosphate • Step 1- Carbamoyl phosphate + ornithine →citrulline (carbamoyl phosphatase 1 • Citrulline + Nitrogen (2nd)→arginino succinate • Arginino succinate → fumarate and arginine • Arginine → urea and regenerate ornithin • Fumarate enter the TCA cycle
Ammonia intoxication • Ammonia produced by enteric bacteria and produced by tissues are rapidly cleared from circulation by the liver and converted to urea • Ammonia is toxic to central nerveous system • Ammonia levels may rise to toxic levels in impaired hepatic function – cirrhosis • Ammonia is toxic to brain – reacts with α-ketoglutarate to form glutamate – depleted levels of α-ketoglutarate – impair fx of TCA cycle in neurons
Ammonia intoxication • Mammals with genetic defects in any enzyme involved in urea formation cannot tolerate protein rich diet- as free ammonia cant be converted to urea- lead to hyperammonemia • Protein free diet is not an option. Mammals are incapable of synthesizing all 20 amino acids, thus must come from diet
Amino acid catabolism • Involve transferring the amino nitrogen to α-ketoglutarate to produce glutamate- leaving behind the carbon skeleton • After removal of their amino groups, the carbon skeleton of aa undergo oxidation to compounds that can enter the TCA cycle • In liver
The fate of carbon skeleton • Glucogenic aa yields pyruvate or OAA on degradation -----> gluconeogenesis • Ketogenic aa breaks down to acetyl-CoA or acetoacetyl-CoA- leading to the formation of ketone bodies
Six aa are degraded to pyruvate • Alanine, tryptophan, cystein, serine, glycine, and threonine • All are converted to pyruvate • Pyruvate can either be converted to acetyl-CoA (Ketone bodies precursor) • Or converted to OAA (gluconeogenesis)
Glycine degradation • Can be degraded in 3 ways – only one yield pyruvate • Involve conversion of glycine to serine via catalysis of serine hydroxymethyltransferase. Serine is then converted to pyruvate
Glycine degradation 2) Glycine undergoes oxidative cleavage to CO2, NH4+ and a methylene group – catalysed by glycine cleavage enzyme (or glycine synthase) • This pathway is critical in mammals • Defects in glycine cleavage enzyme- lead to elevated serum of glycine – inhibit neurotransmitter- mental retardation
Glycine degradation 3) Conversion of glycine to glyoxylate by D-amino acid oxidase. Glyoxylate is further oxidized to oxalate • Crystals of calcium oxalate account for 75% of all kidney stones
Ketogenic amino acid • Phenylalanine, tyrosine, isoleucine, leucine, tryptophan, threonine, and lysine • Breakdown of trp – precursor to synthesis • NAD and NADP in animals • Serotonin – a neurotransmitter in vertebrate
Phenylalanine catabolism • Phenylalanine- precursor for dopamin (a neurotransmitter) and hormones (norepinephrine and epinephrine) • Breakdown of phenylalanine- catalysed by phenylalanine carboxylase • Deficiency of this enzyme lead to phenylketoneuria (PKU) disease – due to elevated levels of phenylalanine
PKU • Defiency of phenylalanine carboxylase cause phenylalanine undergoes transamination with pyruvate – yield phenylpyruvate – • Accumulate in blood and tissues- excreted thru urine • Other than being excreted as urine directly, some of phenylpyruvate are reduced to phenylacetate – give odor to urine – nurses have traditionally used to detect PKU in infants • Accumulation of phenylalanine in early life impairs normal development of the brain- mental retardation • Due to excess of phenylalanine competing with other aa for transport across the blood brain-barrier- deficit required metabolites • Mental retardation can be prevented by rigid diet- provide only enough phenylalanine
The fate of branched amino acid • Isoleucine, leucine, and valine • Degraded only in extrahepatic tissues – oxidized as fuels in muscles, adipose, kidney • Due to the presence of branched-chain α-ketoacid dehydrogenase complex- absent in liver
Maple syrup urine disease • Due to the defective branched-chain α-ketoaciddehydrogenase complex- • Lead to accumulation of leucine in blood- and excreted to urine – smell like maple syrup • Untreated lead to abnormal development of the brain, mental retardation, and death in early infacy • Treatment include limiting the intake of valine, isoleucine, and leucine
Metabolic disorders related to urea cycle • Defects in urea synthesis results in ammonia intoxication • More severe if the metabolic blocks occur at reactions 1 or 2 – some intermediate compound have been synthesized • Due to defects in any several enzyme involved in the cycle