Download

1 / 20
230 likes | 490 Views
Amino Acid Oxidation and the Production of Urea. Lehninger Ch. 18 BIO 322 Recitation 4-2 / Spring 2013. In animals, amino acids undergo oxidative degradation in three different metabolic circumstances :

E N D
Amino Acid Oxidation and the Production of Urea Lehninger Ch. 18 BIO 322 Recitation 4-2 / Spring 2013
In animals, amino acids undergo oxidative degradation in three different metabolic circumstances: • During the normal synthesis anddegradation ofcellular proteins (protein turnover),some amino acids that are released from protein breakdown and are not needed for new protein synthesis undergo oxidative degradation. • When a diet is rich in protein and the ingestedamino acids exceed the body's needs for protein synthes, the surplusis catabolized, aminoacids cannot be stored. • During starvation or in uncontrolled diabetes mellitus, when carbohydrates are either unavailable or not properly utilized, cellular proteins are used as fuel.
Amino acids lose their amino groups to form α-keto acids the carbon skeletons of AA. • Seperation α amino group from carbon skeleton • α-keto acids undergo oxidation to CO2 and H2O, or 3C-4C units that can be converted to glucose via gluconeogenesis
Metabolic Fates of Amino Groups • Nitrogen is abundant in atmosphere but inert for use. Only few organisms can convert nitrogen gas in to useful form ammonia, so amino groups are controlled carefully. • AA from diet are the source of most amino groups. Most of them metabolised in the liver. • As a result : Some generated ammonia is recycled and used for biosynthesis. Excess is excreted directly or converted to urea depending on the organism. • Excess ammonia generated in extrahepatic tissues travel to the liver in the form of amino groups for the conversion to excretory form. • Glutamine and glutamate – collecting point of amino groups • In hepatocytes, amino groups are transferred to α keto glutarate to form glutamate. Then glutamate enters into mitochondria to give the amino group and form ammonia as ammonium ion. • Excess ammonia in other tissue are converted to amide nitrogen of glutamine. • Glutamine and glutamate conc. is higher than other AA. • Skelatal muscle – excess amino groups are transferred to pyruvate to form alanine – another important transport molecule of amino groups to liver
Dietary Protein Is Enzymatically Degraded to AA • Gastric juice : pH 1-2.5 – antiseptic, denaturing agent, increased vulnerability to enzymatic hydrolysis for proteins • Pepsinogen – pepsin at low pH – hydrolyzes peptide bonds on the amino terminal side of aromatic AA Phe, Trp,Tyr – smaller peptide.
Dietary Protein Is Enzymatically Degraded to AA • Trypsinogen, chymotrypsinogen and procarboxypeptidases A and B are secreted from exocrine cells of pancreas. • Trypsinogen to trypsin via enteropeptidase secreted from SI. Free trypsin further catalyzes trypsinogen to trypsin, also activates chymotrypsinogen, procarboxypeptidases and proelastase.
Why this elaborate mechanism for getting active digestive enzymes into the gastrointestinal tract? • Inactive precursors protects exocrine cells from proteolytic attack. • Pancreas protects itself from digestion via pancreatic trypsin inhibitor • İnhibit premature production of active proteolytic enzymes within pancreatic cells. • Pepsin, trypsin and chymotrypsin have different AA specifities. • Degradation of small peptides is completed by other intestinal peptidases such as carboxypeptidase A and B and another aminopeptidase. • Resulting free AA to liver, keratin and some plant protein due to cellulose masking cannot be fully digested. • Acute pancreatitis – active forms of zymogens inside pancreatic cells and attack pancreatic tissue itself pain, organ damage
Pyridoxal Phosphate Participates in the Transfer of α-ketoglutarate • First step in catabolism of L-amino acids in liver is promoted by enzymes called aminotransferases and transaminases. • Transamination reactions, the α amino group is transferred to the α carbon of α- ketoglutarate, leaving behind a α- keto acid. • Aim is to collect amino groups from many different AA in the form of L-glutamate. • Glutamate functions as amino group donor biosynthesis or excretion pathways. • Aminotransferases • Many aminotransferases – specific for α-ketoglutarate but differ in specificity for L-amino acid named for the amino group donor. • Amino transferase rxns are reversible. • Prosthetic group – PLP (pyridoxal phosphate) – coenzyme form of pyridoxine or Vitamin B6
Pyridoxal Phosphate • PLP- Coenzyme of glycogen phosphorylase – not usual role. Primary role is in the metabolism of molecues with amino groups. • PLP- Intermediate carrier of amino groups at the active site of aminotransferases. • PLP- Aldehyde form (pyridoxal phosphate) can accept amino groups and aminated form is called pyridoxamine phosphate, can donate its amino group to α-keto acid • PLP is bound to active site of the enzyme through aldimine (Schiff base) linkage to the ε-amino group of a Lysine residue.
Glutamate Releases Its Amino Group as Ammonia in the liver • Amino groups from many AA are collected in the liver in the form of amino group of L glutamate. • Amino groups from glutamate must be removed to prepare them for excretion. • In hepatocytes: Glutamate to mitochondria – oxidative deamination by glutamate dehdyrogenase. • Present in mitochondrial matrix, only enzyme that can both use NAD or NADP as the acceptor. • Combined action of aminotransferase and glutamate dehydrogenase – TRANSDEAMINATION • α-ketoglutarate from glutamate deamination can be used in TCA cycle for glucose synthesis. • Glutamate dehydrogenrase: Positive modulator ADP, negative modulator GTP • The metabolic rationale has not been elucidated yet. • Mutations at at GTP binding side causes hyperinsulism-hyperammonemia syndrome, characterized by elevated levels of ammonia in the blood stream and hypoglycemia.
Glutamine Transports Ammonia in the Bloodstream • FROM EXTRAHEPATIC TISSUES • Ammonia is toxic to cell. • The free ammonia produced in tissues is combined with glutamate to yield glutamine by action of glutamine synthetase. • Requires ATP. Two step reaction • Glutamate and ATP react to form ADP and gamma-glutamyl phosphate intermediate, which then reacts with ammonia to produce glutamine and inorganic phosphate. • Glutamine is non toxic – Higher concentrations in blood. • Intestine, liver and kidneys • Amide nitrogen is released as ammounium ion in the mitochondria. • Glutaminase catalyzes glutamine to glutamate and ammonia in liver Mitochondria - Ammmonia is converted to urea.
Alanine Transports Ammonia from Skeletal Muscle to the Liver FROM MUSCLE- Muscle amino groups collected as glutamate. Glutamate may be converted to glutamine for transport to liver OR • Glutamate transfers amino group to pyruvate by alanine amino transferase. • Alanine goes to blood, travels to liver. • In cytosol of hepatocytes, alanine amino transferase transfers the amino group of alanine to α -ketoglutarate forming pyruvate and glutamate. • Glutamate goes to mitochondria releases ammonia or can undergo transamination with oxaloacetate to form aspartate, another nitrogen donor in urea synthesis. • Lactate from muscle or erythrocytes to blood, targeted to liver, converted back to glucose – back to muscle (Cori cycle). Energetic burden of gluconeogenesis is imposed on liver rather than muscle
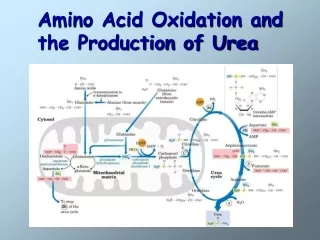
Nitrogen Excretion and The Urea Cycle • In liver mitoch. whatever the source ammonia generated + product of mitochonrial respiration, carbondioxide as bicarbonate, produce carbamoyl phosphate in the matrix by ATP dependent Carbamoyl phosphate synthetase I • Carbamoyl phosphate donates it s carbamoyl group to ornithine to form citrulline, with release of Pi. (Ornithine transcarbamoylase) – Citrulline to cytosol • Ornithine resembles oxaloacetate of TCA cycle beucase it accepts material at each turn of the cycle. • Condensation of amino group of aspartate and ureido (carbonyl) of citrulline forms arginosuccinate. (Arginosuccinate synthetase) – requires ATP, citrullyl-AMP intermediate • Arginosuccinate is then cleaved to form arginine and fumarate Arginosuccinase. Fumarate goes back to mitochondria and joins TCA cycle. – Only reversible step in urea cycle • Cytosolic arginase cleaved arginine to yield urea and ornithine. Ornithine back to mitochonria for another round of urea cycle. • Mitochondrial and cytosolic enzymes are clustered – The citrulline passed directly to the active site of arginosuccinate synthetase chanelling. Only urea is released to general cytosolic pool.
The Citric Acid and Urea Cycles Can Be Linked • Krebs Bicycle - TCA + UREA interconnected. Each can operate independently • TCA enzymes – fumarase and malate dehydrogenase have cytosolic isozymes • Fumarate to malate in cytosol can go back to mitochondria as malate • Aspartate formed in mitochondria via transamination between oxaloacetate and glutamate can be transported back to cytosol where it serves as a nitrogen donor in the rxn catalyzed by arginosuccinate synthetase. • These rxns make up aspartate-arginosuccinate shunt – provide metabolic links
The Activity of the Urea Cycle Is Regulated at Two Levels • When dietary intake is primarly protein- excess AA – excess urea • Prolonged starvation – excess urea due to breakdown of muscle protein • All urea cycle enzymes are synthesized at higher rates in starvation and high protein diet. • Shorter time scale regulation of first enzyme, carbomyl phosphate synthetase I is allosterically activated by N-acetylglutamate, which is synthesized from acetyl-coA and glutamate by N-acetylglutamate synthase. • Arginine, an intermediate of urea cycle is also the activator of N-acetylglutamate synthase.
Humans are incapable of synthesizing half of the 20 common amino acids essential amino acids.
7 amino acids broken down to acetyl-CoA • 5 alfa-ketoglutarate • 4 to succinyl-CoA • 2 to fumarate • 2 to oxaloacetate • 6 to pyruvate acetyl-CoA or oxaloacetate