Download

1 / 43
740 likes | 1.9k Views
Chapter 18 Amino Acid Oxidation and The Production of Urea. Amino Acid Oxidation. Dependency of amino acid as energy source Carnivores> herbivores> microorganism >> plant Amino acid degradation in animals Amino acids for oxidation Extra amino acid during protein turnover

E N D
Chapter 18 Amino Acid Oxidation and The Production of Urea
Amino Acid Oxidation • Dependency of amino acid as energy source • Carnivores> herbivores> microorganism >> plant • Amino acid degradation in animals • Amino acids for oxidation • Extra amino acid during protein turnover • Protein-rich diet (no storage) • During starvation or in uncontrolled diabetes • Removal of amino group (NH4+) • a-keto acid (C skeleton of amino acids) • Oxidation to CO2 & H2O • Sources of C3 or C4 units for gluconeogenesis or fuels
Amino Group Catabolism • Amino acid metabolism amino group (= nitrogen metabolism) • Liver is a major site • Recycle for biosynthetic pathways • Excretion; ammnonia, urea, uric acid • Glutamate & glutamine • General collection point for amino group • NH3 from amino acids + a-ketoglutarate glutamate into mitochondria, release of NH4+ • Source of ammonia • Dietary protein (major source) • Muscle & other tissues NH4+ + glutamate glutamine mitochondria in hepatocytes NH4+ +pyruvate alanine hepatocytes
Digestion of Dietary Protein • In stomach • Entry of diet secretion of gastrin from gastric mucosa secretion of HCl (parietal cells), pepsinogen (chief cells) • Acidic gastric juice (pH 1.0 to 2.5) • Antiseptic & denaturing agent (protein unfolding) • Pepsinogen : zymogen • Conversion to active pepsinby autocatalytic cleavage (at low pH) • Digestion of peptide bonds at Phe, Trp, Tyr mixture of small peptides • In small intestine • Low pH secretion of secretin stimulation of bicarbonate secretion from pancreas neutralization • Arrival in the upper part of intestine (duodenum) release of cholecystokinin into blood stimulation of pancreatic zymogens • Trypsinogen : activated by enteropeptidase • Chymotrypsinogen, procarboxypeptidase A and B : activated by trypsin c.f.) Protection of pancreas from proteolytic digestion • Production of zymogens • Pancreatic trypsin inhibitor • Protein digestion by trypsin, chymotrypsin, carboxypeptidase, aminopeptidase • Uptake of amino acids by the epithelial cells
Digestion of Dietary Protein Blood capillaries Liver
Transamination • 1st step of amino acid catabolism • Transfer of a-amino group to a-ketoglutarate • Generation of L-glutamate & a-ketoacid • Aminotransferase (transaminase) • Amino acid specificity (named after amino acids) • Reversible reaction ; ∆G’° ≈ 0 kJ/mol • Pyridoxal phosphate (PLP) • Bimolecular Ping-Pong reactions
Pyridoxal phosphate (PLP) • Coenzyme form of pyridoxine (vitamin B6) • Intermediate carrier of amino group • Electron sink for carbanion (resonance stabilization) • Transamination • Racemization (L- & D-form interconversion) • Decarboxylation
PLP-mediated transamination at a-carbon PLP-mediated transamination: Ping-Pong mechanism amino acid a-ketoglutarate pyridoxal phosphate pyridoxamine phosphate pyridoxal phosphate a-keto acid glutamate
Oxidative Deamination of Glutamate • Oxidative deamination • Mitochondrial matrix of hepatocytes • Glutamate dehydrogenase • Generation of a-ketoglutarate & ammonia • NAD+ or NADP+ as electron acceptor • Allosteric regulation • By ADP (inhibition) • By GTP (activation) • Transdeamination • Transamination of A.a. + oxidative deamination of Glu • A few amino acids undergoes direct oxidative deamination
Glutamine as Ammonia Carrier in the Bloodstream • Ammonia generated in extrahepatic tissues • Glutamine synthetase • Incorporation of ammonia into glutamate glutamine • Transport of gln to the liver via blood • Higher gln concentration than other amino acids in blood • Glutaminase in the liver, intestine, and kidney • Glutamine Glutamate + NH4+
Alanine Transports Ammonia from Skeletal Muscles to the Liver • Glucose-alanine cycle • In muscle • Glycolysis & degradation of amino acids • Alanine aminotransferase • Transfer amino group of glutamate to pyruvate alanine + a-ketoglutarate • Transport of alanine to the liver • In the liver • Alanine aminotransferase • Transfer amino group of alanine to a-ketoglutarate glutamate + pyruvate • Gluconeogenesis • Pyruvate , lactate glucose • Transport of glucose to muscle
Ammonia is toxic to animals. Comatose state of brain (high brain’s water content) 1. NH3: alkalization of cellular fluid 2. a-ketoglutarate, NADH, ATP: citric acid cycle & ATP production 3. glutamate and GABA (g-aminobutyrate): neurotransmitter depletion
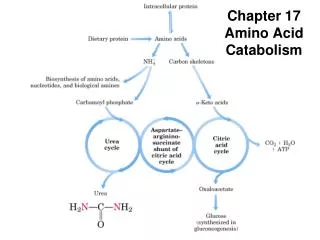
18.2 Nitrogen Excretion and the Urea Cycle Produced in liver Blood Kidney urine
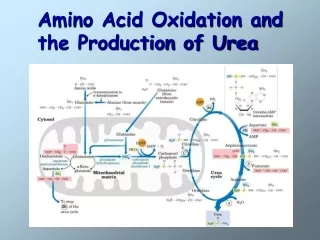
Urea Cycle in Mitochondria • Formation of carbamoyl phosphate; preparatory step NH4+ + HCO3- + 2 ATP carbamoyl phosphate + 2 ADP + Pi Carbamoyl phosphate synthetase I - ATP-dependent reaction • 1st step in the urea cycle; Ornitine + carbamoyl phosphate citrulline + Pi Ornitine transcarbamoylase
Urea Cycle in Cytosol • 2nd step; formation of argininosuccinate Incorporation of the second N from aspartate Argininosuccinate synthetase • ATP requirement • Citrullyl-AMP intermediate • 3rd step; formation of arginine & fumarate Argininosuccinase; only reversible step in the cycle • 4th step; Cleavage of arginine to urea & ornithine Arginase
Asparatate-argininosuccinate shunt • Metabolic links between citric acid and urea cycles • In cytosol • Fumarate to malate citric acid cycle in mitochondria • In mitochondria • OAA + Glu a-ketoglutarate + Asp urea cycle in cytosol • Energetic cost • Consumption • 3 ATP for urea cycle • Generation • Malate to OAA • 1 NADH = 2.5 ATP
Regulation of the Urea Cycle • Long term regulation • Regulation in gene expression • Starving animals & very-high protein diet • Increase in synthesis of enzymes in urea cycle • Short term regulation • Allosteric regulation of a key enzyme • Carbamoyl phosphate synthetase I • Activation by N-acetylglutamate
Treatment of genetic defects in the urea cycle • Genetic defect in the urea cycle • ammonia accumulation; hyperammonemia • Limiting protein-rich diet is not an option • Administration of aromatic acids; benzoate or phenylbutyrate • Administration of carbamoyl glutamate • Supplement of arginine
Amino Acid Catabolism • Carbon skeleton of 20 amino acids • Conversion to 6 major products - pyruvate - acetyl-CoA - a-ketoglutarate - succinyl-CoA - fumarate - oxaloacetate
Glucogenic or Ketogenic Amino Acids • Ketogenic amino acids • Conversion to acetyl-CoA or acetoacetyl-CoA ketone bodies in liver • Phe, Tyr, Ile, Leu, Trp, Thr, Lys • Leu : common in protein • Contribution to ketosis under starvation conditions • Glucogenic amino acids • Conversion to pyruvate, a-ketoglutarate, succinyl-CoA, fumarate, and OAA glucose/glycogen synthesis • Both ketogenic and glucogenic • Phe, Tyr, Ile, Trp, Thr
Enzyme cofactors in amino acid catabolism • One-carbon transfer reactions ; common reaction type, involvement of one of 3 cofactors • Biotin ;one-carbon tranfer of most oxidized state, CO2 • Tetrahydrofolate (H4 folate) ;One-carbon transfer of intermediate oxidation states or methyl groups • S-adenosylmethionine ; one-carbon transfer of most reduced state, -CH3
Tetrahydrofolate • folate (vitamin) to H4 folate • Dihydrofolate reductase • Primary source of one-carbon unit • Carbon removed in the conversion of Ser to Gly • Oxidation states of H4 folate ; One-carbon groups bonded to N-5 or N-10 or both - Methyl group (most reduced) - Methylene group - Methenyl, formyl, formimino group (most oxidized) • Interconvertible & donors of one-carbon units (except N5-methyl-tetrahydrofolate)
S-adenosylmethionine (adoMet) • Cofactor for methyl group transfer • Synthesized from Met and ATP • Methionine adenosyl transferase • Unusual displacement of triphosphate from ATP • Potent alkylating agent • Destabilizing sulfonium ion inducing nucleophilic attack on methyl group
Six amino acids are degraded to pyruvate • Ala, Trp, Cys, Ser, Gly, Thr • pyruvate acetyl-CoA citric acid cycle or gluconeogenesis
3rd pathway of glycine degradation • - D-amino acid oxidase • detoxification of D-amino acid • high level in kidney • - Oxalate crystals of calcium oxalate (kidney stones)
Seven Amino Acids Are Degraded to Acetyl-CoA • Trp, Lys, Phe, Tyr, Leu, Ileu, Thr acetoacetyl-CoA acetyl-CoA
Intermediates of Trp catabolism be precusors for other biomolecules
Catabolic pathways for Phe & Tyr • Phe & Tyr are precusors • dopamine • norephinephrine, epinephrine • melanin
Phenylalanine hydroxylase • Mixed function oxidase • ; Substrate hydroxylation + oxygen reduction to H2O • Tetrahydrobiopterin as a cofactor
Example of A.a. metabolism defects; Phe catabolism • Phedegradaion fumarate + acetoacetyl-CoA • Defects in Phe catabolism • Phenylketonuria (PKU) • Genetic defect in Phe hydroxylase or dihydrobiopterin reductase • Elevated levels of Phe & phenylpyruvate • Mental retardation • Alkaptonuria • Genetic defect in homogentisate dioxygenase • Oxidation of accumulated homogentisate • Black urine • Arthritis
Genetic defects of A.a. metabolism • defective neural development & metal retardation
Five Amino Acids Are Converted to a-ketoglutarate • Pro, Glu, Gln, Arg, His; amino acids with five C skeleton • converging to Glu
Four Amino Acids Are Converted to Succinyl-CoA • Met, Ileu, Thr, Val converging to propionyl-CoA
Branched-Chain Amino Acids Are Not Degraded in the Liver • Leu, Ile, Val • Primarily oxidized as fuels in muscle, adipose, kidney, brain • Branched-chain aminotransferases (not in liver) • Branched-chain a-keto acid dehydrogenase complex