Download
1 / 70
700 likes | 729 Views
Cushing’s disease. Cushing’s disease 70 % of cases F/B = 3 – 8 / 1 20 – 40 years Pituitary Microadenoma : bazohilic sau cromophob, secreting : ACTH, LPH, endorfins 80 –90 % au < 10 mm 50 % < 5 mm (less than 2 mm) Macroadenoma corticotrop cell hyperplasia

E N D
Cushing’s disease • Cushing’s disease • 70 % of cases • F/B = 3 – 8 / 1 • 20 – 40 years • Pituitary • Microadenoma: bazohilic sau cromophob, secreting : ACTH, LPH, endorfins • 80 –90 % au < 10 mm • 50 % < 5 mm (less than 2 mm) • Macroadenoma • corticotrop cell hyperplasia • CRH – secreting gangliocytoma+ corticotroph cell hyperplasia • ADRENALS • Hyperplasia of zona fasciculata and zona glomerularis • in ACTH secreting adenoma may have 8-10 g to 12 –24 g • bilateral nodular hyperplasia – may be initialy ACTH –dependent and than may become autonomous by modifying its ACTH receptors
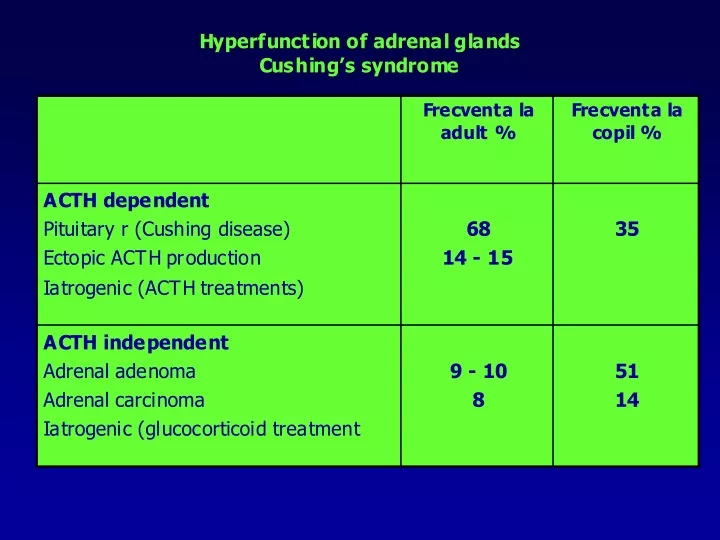
Cushing’s syndrome • ACTH –dependent Cushing’s syndrome due to ectopic ACTH secretion • 15 % din total • M / F = 3 / 1 • 40 – 60 years • ACTH –dependent Cushing’s syndrome due to ectopic ACTH secretion – causes and characteristics • 15 % din total • M / F = 3 / 1 • 40 – 60 years • Small cell lung carcinoma • pancreatic carcinoid tumors • Other carcinoids: lung, small bowel, timus, ovary • Medullary thyroid carcinoma • PHeochromocitom a • Characteristics • Very high level of SCTH, POMC • DXM does not inhibit ACTH and cortisole secretion • it is an important aldosterone and androgen secretion
CUSHING’S DISEASE AND SYNDROME – CLINICAL SIGHNS AND SYMPTOMS
CONGENITAL ADRENAL HYPERPLASIA • They are genetically produced autosomal recessive deficiencies in the steroidogensis in the adrenals with effects on prenatal and /or post natal sexual development. Steroidogeentic defects during fetal life produce cortisole deficiency followed by ACTH increase and adrenal hyperplasia. • Forms: • clasic – obvious at birth • non-clasic – onset during puberty • criptic – detectable only by specific tests • Prenatal sexual development involves the differentiation of the gonads and beginning of their function, (first 2 months), internal genital organs differentiation month 2-3, external genital organs development (months 3-4) brain sexual differentiation (months 6-9). • Postnatal sexual development includes gender identity and gender development, sexual morphology during puberty, sexual behavior. • According with severity of steroidogenic defects, these syndromes are associated with more or less severe sexual development impairment,which may affect entire life.
During critical period of 3-4 months development of external genitalia occurs. They evolve from a common primordium called genital sinus formed of: genital tubercle, urethral folds, genital swelling. • In the presence of testosterone (male sex, testosterone secretion of the testis) the genital tubercle become glans penis urethral folds become penile shaft, and genital swelling fuse on median line to form the scrotum. • In normal female fetus in the absence of androgens genital tubercle becomes gland clitoris, urethral folds remain separate and form labia minora, and genital swelling also separated will form labia majora. • If female fetus has an androgen excess their genitalia will develop more ore less to the male phenotype resulting in ambiguous genitalia. Depending of the severity of androgen excess this ambiguity may evolve from clitoral enlargement to more important male-like appearance. These ambiguities are classified into 5 degrees by Prader (see below). In the same time female brain may suffer androgen imprinting with gender identity and gender role abnormalities. This ambiguities are called female pseudohermphroditism: chromosomal and gonadal sex are those of a female and external genitalia are ambiguous. • If male fetus has no possibilities to produce enough testosterone to produce normal virilisation of external genitalia, these will have the same ambiguities. The situation is called male pseudohermaphroditism – chromosomal and gonadal sex is that of a male but external genitalia are ambiguous. The Prader’s classification of ambiguous genitalia are presented below
Physiopathology of congenital adrenal hyperplasia: • 1. Congenital enzyme deficiency involved in the cortisole synthesis occurs early during fetal life with cortisole deficiency and increased ACTH secretion that will result in adrenal hyperplasia • 2. Sterodogenesis in strongly stimulated with increased production of sterodogenetic products situated before the affected enzyme and of those products that are formed within the unaffected steroidogenetic chain. • 3. Steroidogenetic products situated before affected enzyme will accumulate in large concentration in fetal and child blood and will be excreted in urine allowing the diagnosis of sterodogenetic defect. • 4. If sterodogenesis is unaffected in the androgen biosynthesis chain fetuses of both sexes will have increased concentration of androgens that will result in virilization female external genitalia ( female pseudo hermaphroditism) in female fetus and newborn. • 5. If steroidogenesis of androgens is affected , this event will occur also in the testes and ovaries. According to the degree of androgen deficiency male genitalia will be undervirilized or will have a female aspect. – male pseudo hermaphroditism. During puberty and adulthood testosterone and oestradiol synthesis will be impaired. • During childhood CAH with androgen excess will produce further virilization in females resulting a heterosexual pseudoprecoccious puberty in girls at 5-6 years (muscle development, acne, sexual hair growths, deepening of voice, absence of breast development and precocious fusion of epiphiseal growth plates with impaired final height). and isosexual psudoprecoccious puberty in boys, 5-6 years (muscle development, excessive sexual hair growth, deepening of the voice, small testes) . • In CAH forms which affect testosterone synthesis puberty does not occur in both sexes. • Metabolic abnormalities depend of cortisole deficiency and aldosterone deficiency or excess according to the affected enzyme



















