Download
1 / 35
410 likes | 872 Views
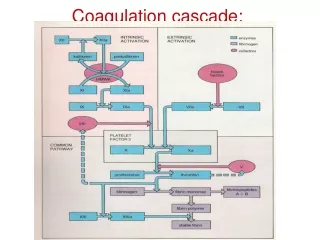
Coagulation. Janell Wilcox, PA-C. Hemostasis. Definition: clotting of the blood Phases Formation of the platelet plug Coagulation cascade Termination of clotting by antithrombotic control mechanisms Removal of the clot by fibrolysis. Clotting diagram.

E N D
Coagulation Janell Wilcox, PA-C
Hemostasis • Definition: clotting of the blood • Phases • Formation of the platelet plug • Coagulation cascade • Termination of clotting by antithrombotic control mechanisms • Removal of the clot by fibrolysis
Clotting diagram • Courtesy of: Essential Haematology, 6th Ed. – Hoffbrand & Moss
Platelet plug Starts with vessel injury! • Activation • Collagen impairment and thrombin activation • Adhesion • Deposition of platelets on the subendothelial matrix • Shape change causing platelets to be extremely adhesive • Aggregation: Platelet-platelet cohesion • Leading to binding of both immobilized VWF and fibrinogen • Secretion • Release of platelet granule proteins resulting in stabilization and further platelet aggregation. • Procoagulant activity – Enhancement of thrombin generation; ultimate assembly of enzyme complexes in the clotting cascade on the platelet surface.
Vasoconstriction • Vessel injury results in vasoconstriction and reduced blood flow helping to form the primary hemostatic plug
Extrinsic pathway • Definition: activated by tissue factor exposed at the site of injury or tissue factor-like material (thromboplastin) • Lab test: Prothrombin clotting time (PT) • Vessel wall damage leads to expression of tissue factor (tissue throboplastin) - normally exposed to blood flow only after endothelial damage • Activation of VII, IX (Extrinsic) and • X (Common Pathway)
Intrinsic Pathway • Initiated by the exposure of blood to a negatively charged surface. • This activates several plasma proteins (Most notable Factor XII; also prekallikrein (fletcher factor) and high molecular weight kininogen (fitzgerald factor) • activating these proteins activates XI which activates factor IX • IXa in complex with factor VIIIa forms intrinsic X-ase leading to the formation of factor Xa and thrombin • Resulting in an increase in factor VIII activation as factor Xa and thrombin are formed. • Lab test: activated partial thromboplastin clotting time (aPTT)
Common pathway • Both pathways converge on the activation of factor X – job is to convert prothrombin to thrombin (the final enzyme of the clotting cascade) • Thrombin converts fibrinogen from soluble plasma protein in to an insoluble fibrin clot. • Dual pathway needed because there is limited amount of tissue factor generated, thus sustained generation of thrombin depends on activation of factor IX.
Termination of clotting • We need blood flow! • Termination phase includes: • Two circulating enzyme inhibitors: • Antithrobmin: • neutralizes enzymes of the clotting cascade • tissue factor pathway inhibitor: • inhibits factor X • Protein C and S pathway – • inactivates factors V and VIII • AND prostacyclin, thromboxane, nitric oxide, and platelet reactivity
Clot elimination and Fibrinolysis Must restore vessel patency • Clot must be oraganized and removed • Process: • Plasminogenbinds fibrin and tissue plasminogen activator (tPA) • THEN activate proteolyticplasmin • THAT cleaves fibrin, fibrinogen and a variety of plasma protein and clotting factors
Platelet disorders • Thrombocytopenia • Under-produced • Increased destruction • Thrombocytosis • Qualitative platelet disorders
Thrombocytopenia - underproduction • Most common cause of thrombocytopenia • Causes • Drug induced – iatrogenic – we cause it! • Chemotherapy, radiation, ethanol • Viral suppression • Many common viruses and HIV • Marrow infiltration • Tumors, storage diseases or infectious agents • Leukemia, multi-myeloma,
Thrombocytopenia – with increased destruction • ITP (Autoimmune (ideopathic) thrombocytopenic purpura) • Post-transfusion purpura (very rare) • Drug-induced immune throbmocytopenia • TTP (thrombotic thrombocytopenia purpura (TTP) / hemolytic-uremic syndrome (HUS), new term TMA (thrombotic microangiopathic anemia) • Thrombocytopenia during pregnancy • Hyperslenism
ITP • Relatively common disorder (most common without anemia or neutropenia) • May be associated with systemic lupus, HIV, h. pylori, CLL, hodgkin’s lymphoma, or autoimmune haemolytic anemia • Pathology: platelet autoantibodies result in premature removal of platelets from circulation • Clinical features: Insidious onset of petechialhemorrhage (new Rash – flat tiny dotted bruises – come on with low platelet count), easy bruising, and menorrhagia. Mucosal bleeding in severe cases. • Diagnoses: Low Platelet count 10-100,000 (normally 150,000), reduced number of platelets on smear, increased megakaryocytes on bone marrow, sensitive tests of antiglycoprotein (GPIIb/IIIa on platelet surface) • Treatment: corticosteroids, Immunoglobulin, immunosuppressive drugs, monoclonal antibody, thrombopoietin-receptor agonist, splenectomy.
Post transfusion purpura • Rare • Occurs about 10 days after transfusion • Caused by antibodies in recipient developing against human platelet antigen-1a on transfused platelets • Cause is unknown • Treatment: immunoglobulin, plasma exchange, or corticosteroids
TTP/HUS • Deficiency of the ADAMTS13 metalloprotease which breaks down ultra large vanwilleand factor multimers • Familial forms there are more than 50 ADAMTS13 mutations • Diagnosis • Pentad: Thrombocytopenia, microangiopathic hemolytic anemia, neurologic abnormalities, renal failure, and fever • Look out for: Severely Elevated LDH (normal 100-200 – it will be around 4,000) and schistocytes (broken RBC little crescent shaped RBC) • Look for renal failure • 80% mortality rate • Treatment: • Plasma exchange, FFP, and cryopersipitate. Do not transfuse platelets • Stay away from platelets, can create a negative feedback making problem worse. • Infection – Most common causeGet rid of cause with a plama exchange. If they bleed, give cryopersipitate and FFP as they have clotting factors within them
DIC Wide spread inappropriate intravascular deposition of fibrin with consumption of coagulation factors and platelets caused by increased activity of thrombin in the circulation that overwhelms its normal rate of removal • Clinical features: bleeding from venepunctures or wounds, GI, oropharynx, lungs, vaginal. Less frequent: microthrombi cause skin lesions, renal failure and gangreen • Lab: Low platelet count, Fibrinogen concentration is low, Thrombin time is prolonged, high d-dimer, PT and aPTT are both elevated • Smear: Hemolytic anemia (schistocytes – broken RBCs) • Causes: Infection, malignancy, obstetric complications, hypersensitivity reactions • Treatment: Treat underlying cause! • Bleeding – supportive therapy with FFP and cryoprecipitate • Try to stay away from platelets • Thrombosis – heparin or antiplatelet drugs
Increased Splenic pooling • In Splenomegaly • Malignancy: CLL Chronic Lymphocidic Leukemia • up to 90% of the platelets may be sequestered in the spleen • Normal: • only 1/3 of total platelets of the body should be in spleen. If 90% of them are there, it’s bad • Not usually associated with bleeding • May require a splenectomy
Qualitative dysfunction (Disorders of platelet function) • Hereditary: Very, Very Rare • Thrombasthenia (Glanzmann’s disease) • Bernard-Soulier syndrome • Storage disease • Acquired disorders • Caused by Antiplateletdrugs (aspirin and ethanol – Affect platelet function) • Hyperglobulinemia • Uremia: High BUN, platelets still work • Myeloproliferative and myelodysplasticdisorders • Myeloproliferative – listen up • Myelodysplastic – precursor to leukemia
Thromobcytosis – Too many platelets • Reactive throbcytosis – iron deficiency, chronic infection, inflammatory conditions, solid tumors, after splenectomy • No treatment usually needed • Essential thrombocythemia (ET) – • diagnosis of exclusion with platelet count greater than 600,000. Send for hematologist treatment
Acquired disorders of coagulation • Vitamin K deficiency – Very Common… • malabsorption states, poor dietary intake • Diagnosis: PT and PTT are prolonged • Treatment: Vitamin K po or IV or rapid correction with prothrombin complex concentrate (for a few days to normalize PT and PTT). Rapid correction via FFP or cryoprecipitate as they have clotting factors built in). • Hemorrhagic disease of newborn • Baby liver is underdeveloped. PT/PTT are produced there. May have problem with bleeding. • Liver disease • PT/PTT are produced there. If not working properly, you will bleed • If PT/PTT/INR is elevated, liver is not working properly • Elevated AST, ALT, Billurubin as well • DIC – • correct the underlying cause (sepsis, malignancy, trauma) • Acquired inhibitors of coagulation are seen in hemophilia, postpartum with underlying immunologic disease, or in otherwise normal individuals.
Inherited bleeding Disorders • Hemophilia: • Hemophilia A – deficiency in factor VIII the ciritical cofactor in generation of facterXa by factor Ixa • Hemophilia B – Factory IX deficiency • VonWillebrand’s Disease – most common inherited bleeding disease • Numerous mutations in the large vWF gene resulting in a spectrum of disease • Treatment: DDAVP, plasma product infusion.
Hemophilia A Absence or low levels of plasma Factor VIII. Gene is on X chromosome • Clinical features: Profuse post circumcision hemorrhage, joint / soft tissue bleeding, excessive bruising, recurrent painful hemarthoroses, muscle hematomas leading to progressive joint deformity and disability • Lab: Increased aPTT and factor VIII clotting assay • Treatment: Factor VIII level replacement and / or DDAVP
Hemophilia B (Christmas disease) • Factor IX deficiency • Lab: aPTT elevation and FactorIX clotting assay abnormalities • Treatment: Factor IX
VonWillebrand disease • Reduction in or abnormal function of VonWillebrand factor • Clinical features: Mucous membrane bleeding, excessive blood loss from superficial cuts, abrasions, operative, and post-traumatic hemorrhage • Lab: PFA-100 test abnormal, Factor VIII levels is low, aPTT prolonged, VWF is low, defective platelet aggregation, collagen–binding function reduced, multimer analysis for diagnosing different subtypes • Treatment: Antifibrinolytic agents, DDAVP, VWF concentrates
Venous thrombosis (DVT) • Virchow’s Triad – Describes clot potential • Alteration in blood flow • Vascular endothelium injury • Alterations in constituents of blood • (inherited or acquired hypercoagulable state)
Thrombosis • Superficial venous thrombosis • Red, Painful, go away on there own with hot compress. Don’t move to heart • Inherited thrombophilia • Deep vein thrombosis / pulmonary embolism • DVT • Acquired thrombophilia • Problem with too much clotting
Inherited thrombophilia • Factor V leiden gene mutation: most common inherited cause of increased risk of venous thrombosis:Failure of protein C activation • Women with multiple miscarriages, look at these! • Antithrombin deficiency • Protein C deficiency • Protein S deficiency • Hyperhomocysteinemia • – may be genetic or acquired
DVT risk factors • Surgery (ortho) • Previous DVT • Immobility • Malignancy • Inflammation • Blood disorders • Estrogen therapy • Antiphospholipid syndrome • Increased risk of clotting from inherited diseases in prevous slide. Lots of mis-carriages or multiple clots
DVT: Signs and symptoms • Calf swelling or tenderness, red • Pitting edema • Collateral superficial non-varicose vein • Homan’s sign (pain in calf on flexing of ankle) • Not always +, can’t exclude clot • Wells score clinical assessment tool: point for active cancer, paralysis, bed more then 3 days, surgery within 4 weeks, entire leg swollen, pitting edema, collateral veins • Low probability 0-1 • High probability 2 • Test for with • Ultrasound. • D-Dimer may be high • PE concern – get CT with contrast or VQ scan
Treatment - anticoagulation • Fibrinolytic: Lysis fresh thrombis • Streptokinase and tissue plasminogen activator Used for large clot or PE • Inferior vena cava filters • For cancer patients, who can’t do long term anticoagulation • Standard therapy • IV: Start with heparin or LMWH then change to po to go home • Heparin • Risk of HIT (heparin induced thrombocyopenia) • Low molecular weight heparin • Oral: Warfarin(Coumadin is a Vitamin K antagonists) • Target INR: • 2-3 for 1st DVT • 2.5-3.4 for recurrent DVT while on Warfarin
Length of Treatment • First thormboembolic event in context of a reversible or time-limited risk factor: 3 months • Surgery, in bed for 3-6 months • First ideopathic(don’t know why they had event) event: minimum of 3 months followed by evaluation for risk / benefit ratio of long-term therapy • Patients that may have inherited disorders (factor V) • First unprovoked episode of proximal DVT: indefinite therapy • Close to heart and lungs, higher risk for problems • Malignancy related: treat clot until cancer resolves • Treat until cancer is gone (never less than 6 months)
What you should know now • Formation of a platelet plug • Clotting cascade • Platelet disorders • Coagulation disorders • Clotting – not enough ortoo much
References • Up to Date • Essential Haematology 6th edition, A.V. Hoffbrand, P.A.H. Moss • Hematology and Oncology Subspecialty Consult Second Edition • Christopher Wilcox, MS in Information Technology