Download

1 / 29
310 likes | 755 Views
PORFIRIE DEFINIZIONE BIOSINTESI DELL’EME E REGOLAZIONE EZIOLOGIA, PATOGENESI CLASSIFICAZIONE PORFIRIE COMUNI PORFIRIE RARE. PORFIRIE gruppo di malattie metaboliche rare, per lo più ereditarie causate dalla ridotta attività di uno degli enzimi della via biosintetica dell'eme

E N D
PORFIRIE DEFINIZIONE BIOSINTESI DELL’EME E REGOLAZIONE EZIOLOGIA, PATOGENESI CLASSIFICAZIONE PORFIRIE COMUNI PORFIRIE RARE
PORFIRIE • gruppo di malattie metaboliche rare, per lo più ereditarie • causate dalla ridotta attività di uno degli enzimi della via biosintetica dell'eme • determinano livelli eccessivi di varie sostanze dette porfirine o dei loro precursori nella via metabolica • che si accumulano in sedi diverse • vengono escreti nelle urine e nelle feci • determinano manifestazioni patologiche per i loro effetti su sistema nervoso e cute
Le porfirie sono malattie genetiche multifattoriali in cui fattori ambientali e fisiologici interagiscono per causare le manifestazioni patologiche. Tra questi: • ormoni steroidei • farmaci • stato di nutrizione • I sintomi delle porfirie sono simili a quelli di molte altre malattie maggiormente frequenti, per cui la diagnosi è spesso tardiva. La diagnosi di porfiria deve essere sospettata ed esclusa più frequentemente di quanto venga confermata.
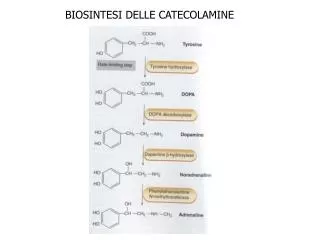
BIOSINTESI DELL’EME L’EME è un pigmento contenente ferro, incluso nelle emoproteine (emoglobina, citocromi, mioglobina) La sua sintesi avviene in 8 tappe, la prima e le ultime tre nel mitocondrio, le altre nel citosol
1. ALA sintetasi • Condensa glicina e succinil • CoA in presenza di vit B6 • Geni diversi per l’enzima • eritroide ( anemia sideroblastica • X-linked) e non eritroide • nel fegato l’enzima è • limitante e viene indotto da • steroidi e farmaci • 2. ALA deidratasi • condensa 2 ALA in PBG • in una reazione con Zn • inibita da Pb • inibita da succinillattone • un analogo dell’ALA • presente in tirosinemia • ereditaria • 3. HMB sintetasi • (PBG deaminasi) • condensa 4 PBG in HMB • 1 gene per 2 isoenzimi • (eritroide e non) • per alternative splicing • inibita da succinillattone • 8. FERROchelatasi • inserisce il ferro nella • protoporfirina • 4. URO III sintetasi • chiude la molecola • dopo inversione anello D • se deficitario la molecola • si chiude senza inversione • e forma URO I, precursore • di COPRO I • 7. PROTO ossidasi • produce una porfirina, • forma ossidata del • porfirinogeno • 5. URO decarbossilasi • agisce in sequenza sui 4 • gruppi carbossimetilici di • URO III per dare COPRO III • agisce anche su URO I per • dare COPRO I • 6. COPRO ossidasi • trasforma i gruppi propionici • In vinilici per dare proto- • porfirinogeno • non agisce su COPRO I
CONTROLLO DELLA BIOSINTESI DELL’EME • 85% prodotto nelle cellule eritroidi per formare emoglobina: • l’ALA sintetasi eritroido-specifica è regolata dal ferro • 10-15% prodotto nel fegato per formare citocromi (P450): • il passaggio limitante è al livello dell’ALA sintetasi, indotto da ormoni e farmaci, e inibito da eme libero
EZIOPATOGENESIdipende da deficit enzimatici ANEMIA SIDEROBLASTICA PORFIRIE
PATOGENESI • ogni porfiria si associa a un deficit enzimatico, con notevole eterogeneità a livello molecolare • il deficit causa l’ accumulo di precursori che si riversano nel sangue e si accumulano in altri tessuti e vengono escreti in urine e feci • se si accumulano precursori precoci (ALA e PBG) si hanno sintomi neurologici: DOLORE ADDOMINALE e DEBOLEZZA MUSCOLARE con paralisi • se si accumulano porfirine si ha fotosensibilità con lesioni cutanee
CLASSIFICAZIONE Esistono diversi tipi di classificazione: Deficit enzimatico: Origine dei precursori in eccesso: Porfirie epatiche Porfirie eritropoietiche Caratteristiche cliniche: Porfirie acute Porfirie cutanee
PORFIRIA ACUTA INTERMITTENTE • autosomica dominante, dovuta a deficit di PBG deaminasi (50% riduzione dell’attività in tutti i tessuti) • incidenza 5/100,000 (più elevata nelle popolazioni psichiatriche) • manifestazione clinica dopo la pubertà, prevalentemente nelle donne (seconda metà del ciclo mestruale) • può essere disabilitante, raramente fatale • per lo più latente, in una minoranza di casi si manifesta con attacchi • FATTORI PRECIPITANTI: (inducono ALAsintetasi epatica ed enzimi contenenti citocromo P450) • Alimentazione: diete a basso contenuto calorico e glicidico, alcool • Ormoni: progesterone e steroidi • Stress chirurgico, infettivo • FARMACI
Sintomi e segni:accessi che si sviluppano nell'arco di ore o giorni e possono persistere per giorni, settimane • sintomi sono dovuti agli effetti sul sistema nervoso; la cute non viene interessata • Le manifestazioni addominali sono dovute agli effetti sui nervi viscerali. • dolore addominale, il sintomo più comune, può essere così intenso da simulare un addome acuto. • nausea, vomito, stipsi e diarrea. • distensione addominale dovuta a ileo paretico. • Con lo stesso meccanismo può essere coinvolta la vescica • ritenzione urinaria, incontinenza, disuria e pollachiuria.
Reperti obiettivi spesso scarsi rispetto alla gravità dei sintomi. Dal momento che non è presente un'infiammazione, la dolorabilità palpatoria addominale e la dolorabilità di rimbalzo non sono particolarmente pronunciate e la temperatura corporea è normale o solo modicamente aumentata. • Tachicardia, ipertensione, sudorazione profusa e irrequietezza (effetti sul sistema nervoso autonomo e agli eccessivi livelli ematici di catecolamine). • neuropatia motoria, specialmente in coincidenza con attacchi gravi o prolungati, ed essa è indicativa di un danno agli assoni dei neuroni motori • debolezza muscolare generalmente inizia alle spalle e alle braccia e può coinvolgere ogni motoneurone, compresi i nervi cranici. Si possono verificare paralisi gravi, insufficienza respiratoria e, raramente, la morte. • tremori e convulsioni • sintomi psichiatrici come l'agitazione e le allucinazioni. • ritenzione idrica e iponatriemia da inappropriata secreazione di ADH, presumibilmente dovuta al coinvolgimento dell'ipotalamo, può portare a o può contribuire all'insorgenza delle convulsioni.
Terapia ricovero ospedaliero ricerca di complicanze neurologiche. gli attacchi gravi vengono trattati con eme per via endovenosa (3 mg/ kg per 4 giorn)i. I sintomi regrediscono entro alcuni giorni. Se la terapia con eme viene ritardata, il danno nervoso progredisce verso stadi più avanzati e il recupero è più lento e può essere incompleto. La terapia con eme deve essere iniziata precocemente, ma solo dopo che la diagnosi di un accesso di porfiria viene confermata mediante la dimostrazione di un marcato aumento del PBG urinario. La diagnosi diventa più difficile per diversi giorni dopo l'inizio della terapia, a causa della pronta riduzione dei livelli di PBG. Terapia sintomatica: Il dolore viene controllato con gli analgesici narcotici. La nausea, il vomito, l'ansia e l'agitazione vengono trattati con dosi di fenotiazine da piccole a moderate. Per l'insonnia benzodiazepine a durata d'azione breve a basse dosi La distensione vescicale può richiedere la cateterizzazione La terapia degli attacchi convulsivi è problematica, perché praticamente tutti i farmaci anticonvulsivanti (eccetto i bromuri e forse la gabapentina) possono esacerbare le porfirie acute. Le convulsioni che compaiono durante gli accessi della malattia possono essere la conseguenza diretta della porfiria oppure possono essere provocate dall'iponatriemia. In alcuni pazienti, porfiria e convulsioni idiopatiche coesistono. Se si ritiene che le convulsioni siano legate alla presenza di un attacco acuto, gli anticonvulsivanti possono essere sospesi una volta ottenuta la risoluzione del quadro.
Prevenzione • i familiari devono essere sottoposti a screening per identificare i casi latenti e istituire misure preventive • Devono essere evitati i farmaci dannosi. • Devono essere evitate le diete drastiche e anche brevi periodi di digiuno (p. es., dopo gli interventi chirurgici o nel corso di malattie intercorrenti). I regimi alimentari per l'obesità devono assicurare una perdita di peso graduale durante le fasi di remissione clinica della porfiria. • La terapia con eme può prevenire la recidiva frequente degli accessi, ma non esistono in proposito regimi posologici standardizzati. Attualmente gli studi suggeriscono che una singola infusione di eme una volta o due alla settimana può essere efficace. • Gli attacchi frequenti in periodo premestruale possono essere prevenuti con l'impiego associato di un analogo dell'ormone di rilascio delle gonadotropine e di una terapia estrogenica sostitutiva a basse dosi, anche se questo genere di utilizzo è ancora in fase sperimentale. Talvolta vengono impiegati con successo i contraccettivi orali, ma esiste il rischio che il progestinico aggravi la porfiria.
PORFIRIA CUTANEA TARDA Il segno caratteristico della porfiria cutanea tarda (PCT) è la fotosensibilità, che provoca la formazione cronica di vesciche a carico della cute esposta alla luce solare. La maggior parte dei pazienti con PCT sembra non avere mutazioni a livello del gene della uroporfirinogeno decarbossilasi. L'enzima è notevolmente ridotto, ma solo nel fegato, in questi pazienti, i quali vengono a volte classificati come affetti da PCT di tipo I. Il deficit dell'enzima epatico è probabilmente acquisito, sebbene il meccanismo non sia stabilito e una base genetica sia comunque possibile. Una minoranza di pazienti (fino al 20%) ha un deficit ereditario di uroporfirinogeno decarbossilasi, che porta l'enzima a essere circa la metà del normale in tutti i tessuti fin dalla nascita. Sebbene questi soggetti vengano classificati come affetti da PCT di tipo II, dal punto di vista clinico essi non sono diversi dai pazienti con il tipo I, salvo per il fatto che l'esordio della sintomatologia può essere più precoce e che altri familiari possono essere stati colpiti dalla stessa malattia. I medesimi fattori precipitanti sono importanti sia nel tipo I sia nel tipo II e la terapia è la stessa. L'attività dell'uroporfirinogeno decarbossilasi deve essere notevolmente inferiore alla metà del normale nel fegato, prima che il tipo I o il II della PCT si manifestino clinicamente. In aggiunta al deficit di uroporfirinogeno decarbossilasi, l'ossidazione dei substrati di questo enzima (uro-, epta-, esa- e pentacarbossilporfirinogeni) che dà luogo alle loro corrispondenti porfirine può essere un meccanismo che contribuisce all'insorgenza della PCT.
Fattori precipitanti • ferro (in quantità normali o aumentate) • l'alcool • virus dell'epatite C • estrogeni • occasionalmente gli idrocarburi clorurati (p. es., l'esaclorobenzene), fumo, 'HIV. • Questi fattori, soprattutto in combinazione con il ferro, possono causare la formazione a livello epatico di specie reattive dell'ossigeno responsabili del danno, le quali inattivano l'uroporfirinogeno decarbossilasi oppure ossidano il suo substrato. Enormi quantità di porfirine si accumulano progressivamente nel fegato e vengono trasportate nel plasma fino alla cute.
Sintomi e segni Sulle aree esposte al sole come il volto, le braccia e il dorso delle mani si formano vescicole e bolle. La cute, soprattutto quella delle mani, appare inoltre fragile e vulnerabile ai minimi traumi. A queste alterazioni fa seguito la formazione di croste e cicatrici. La cicatrizzazione è lenta e spesso seguita da iper- e ipopigmentazione, ipertricosi (soprattutto facciale) e alterazioni pseudosclerodermiche. Generalmente si sviluppa un danno epatico, il quale può essere in parte dovuto alle porfirine, all'infezione cronica da virus dell'epatite C o all'abuso di alcol. Il quadro istopatologico epatico comunemente comprende siderosi, steatosi, necrosi e modificazioni infiammatorie croniche. Alla fine si possono sviluppare cirrosi e carcinoma epatocellulare. I depositi di ferro sono normali o aumentati nella PCT. I soggetti eterozigoti od omozigoti per l'emocromatosi familiare possono avere una predisposizione nei confronti della PCT.
Diagnosi Le lesioni cutanee croniche vescicolari e crostose a carico delle zone esposte alla luce solare sono caratteristiche della PCT. Le biopsie cutanee sono di supporto alla diagnosi, ma non sono specifiche. Altre porfirie (specialmente la porfiria variegata) possono causare lesioni identiche. Alcuni farmaci e sostanze fotosensibilizzanti sconosciute possono causare una pseudoporfiria le cui lesioni somigliano a quelle della PCT, ma in questo caso le porfirine non sono aumentate. Le analisi delle porfirine sono essenziali per la diagnosi: tutte le porfirie che causano lesioni cutanee presentano elevati livelli plasmatici di porfirine. Nella PCT, le porfirine sono aumentate anche nelle urine e in minor misura nelle feci, con un pattern caratteristico. Le porfirine presenti nelle urine sono per lo più uroporfirine ed eptacarbossilporfirine, con un aumento minore delle coproporfirine e delle 5-e 6-carbossilporfirine. Piccole quantità di isocoproporfirine possono essere identificate nel siero o nelle urine, ma nelle feci esse rappresentano spesso le porfirine predominanti; l'aumento delle isocoproporfirine nelle feci (o un aumento del rapporto isocoproporfirine/coproporfirine) è praticamente diagnostico di PCT. Queste tetracarbossilporfirine inusuali si formano quando il pentacarbossilporfirinogeno si accumula a causa del deficit di uroporfirinogeno decarbossilasi e viene quindi parzialmente metabolizzato dalla coproporfirinogeno ossidasi formando isocoproporfirinogeno. La PCT di tipo II viene diagnosticata sulla base del reperto di una riduzione dell'uroporfirinogeno decarbossilasi negli eritrociti, associata a un pattern di eccesso di porfirine caratteristico della PCT. Questo enzima deve essere dosato prima di eseguire la flebotomia, perché l'aumento dell'eritropoiesi può aumentare la quantità di enzima presente negli eritrociti e rendere più difficoltosa l'identificazione di un deficit. Il riscontro di una riduzione dell'attività enzimatica non modifica il trattamento. Sono state descritte famiglie con apparente ereditarietà per PCT che avevano un'uroporfirinogeno decarbossilasi eritrocitaria normale; questo fenomeno viene a volte classificato come PCT di tipo III, ma la natura del difetto genetico non è conosciuta.
Terapia La PCT è la porfiria più facilmente trattabile. L'identificazione e l'allontanamento dei fattori precipitanti sono i primi provvedimenti da prendere, anche se il beneficio clinico è inconsistente. La flebotomia è solitamente efficace nell'induzione delle remissioni cliniche ed è la più diffusamente raccomandata. Viene rimosso circa mezzo litro di sangue ogni 1 o 2 settimane e solitamente sono necessari solo da 5 a 6 salassi. Questo provvedimento impoverisce il fegato di ferro inducendo nel paziente una lieve carenza marziale. Quando il livello sierico della ferritina (un indicatore dei depositi corporei di ferro) scende lievemente al di sotto della norma, la flebotomia viene interrotta. In seguito a salassi eccessivi può svilupparsi anemia. Esistono prove convincenti che gli effetti benefici della flebotomia derivano dalla riduzione dei depositi corporei di ferro. Le porfirine plasmatiche e urinarie diminuiscono gradualmente con il trattamento, un po' in ritardo ma parallelamente alla diminuzione della ferritina. Le lesioni cutanee migliorano e alla fine scompaiono. Dopo la remissione, non è necessario proseguire il trattamento o mantenere bassi i livelli di ferritina. Ulteriori salassi si rendono necessari solo nel caso di una recidiva. L'astinenza dall'alcol aiuta a mantenere lo stato di remissione. La terapia estrogenica può essere ripresa dopo la flebotomia qualora sia di beneficio, come nelle donne in post-menopausa, e raramente provoca recidive. La clorochina o l'idrossiclorochina a basse dosi, metà di una compressa standard (corrispondente rispettivamente a 125 o 100 mg PO due volte alla settimana), costituiscono un'utile alternativa quando il salasso non è praticabile. Questi farmaci rimuovono l'eccesso di porfirine dal fegato. Dosi più alte rimuovono le porfirine troppo rapidamente, causando un peggioramento transitorio della porfiria e un danno epatico. Il regime a basse dosi può essere interrotto dopo aver ottenuto la remissione. L'efficacia della terapia con clorochina e quella della flebotomia sono probabilmente sovrapponibili. Sia la forma familiare sia quella non familiare della PCT rispondono bene sia al salasso sia alla clorochina a basse dosi, ma per altri tipi di porfiria non è così. Pertanto, è fondamentale porre una diagnosi accurata prima di dare inizio al trattamento.
PROTOPORFIRIA ERITROPOIETICA Malattia autosomica dominante, è la forma più comune di porfiria eritropoietica e probabilmente la terza porfiria in ordine di frequenza ed è la conseguenza di un deficit di ferrochelatasi Nella protoporfiria eritropoietica (Erythropoietic ProtoPorphyria, EPP), un deficit di ferrochelatasi determina un aumento della protoporfirina nel midollo osseo e negli eritrociti. Questo aumento ne causa l'ingresso nel plasma e l'escrezione nella bile e nelle feci da parte del fegato. La malattia è caratterizzata dalla comparsa di fotosensibilità cutanea in età infantile, che si manifesta principalmente con dolore, arrossamento e gonfiore che fanno immediatamente seguito all'esposizione alla luce solare. La prevalenza della EPP non è stata calcolata definitivamente. Non esistono predisposizioni di razza o di sesso. Una forma bovina di EPP viene trasmessa come carattere autosomico recessivo. In famiglie diverse affette da EPP sono state identificate molte mutazioni differenti a carico del gene per la ferrochelatasi. La gravità della malattia varia notevolmente tra i diversi pazienti; questa variabilità si osserva anche nell'ambito della stessa famiglia, nella quale più individui hanno ereditato la medesima mutazione ma alcuni hanno un aumento minimo o nullo della protoporfirina eritrocitaria. Pertanto, la variabilità della gravità della EPP non viene spiegata del tutto dalla natura delle differenti mutazioni. Piuttosto, essa può essere dovuta a un tratto genetico che determina una sottoespressione del gene normale per la ferrochelatasi ereditato dall'altro genitore. Come conseguenza, i pazienti con manifestazioni cliniche di EPP possono avere un'attività della ferrochelatasi inferiore a quella che ci si aspetterebbe da una semplice ereditarietà di tipo autosomico dominante (pari alla metà dell'attività normale).
Sintomi e segni I sintomi generalmente hanno un esordio precoce nel corso della vita. Subito dopo l'esposizione alla luce solare (talvolta dopo pochi minuti) si sviluppano dolore urente, prurito, eritema e un gonfiore che può somigliare all'angioedema. Questa sintomatologia non è caratteristica di altre porfirie. Con l'esposizione frequente al sole, possono verificarsi indurimento e ispessimento della cute del dorso delle mani, lievi cicatrici e alterazioni delle unghie, ma vesciche e cicatrici profonde non sono usuali. Poiché i disturbi soggettivi sono maggiori del danno cutaneo obiettivo, la malattia può non essere riconosciuta anche in pazienti con sintomi gravi. I pazienti possono sviluppare calcoli della colecisti contenenti protoporfirina, conseguenza dell'elevata concentrazione di protoporfirina nella bile. La malattia può presentarsi anche con un'inspiegata, lieve alterazione delle prove di funzionalità epatica. L'epatopatia cronica, sebbene infrequente, può essere grave. L'insufficienza epatica si verifica di rado, ma può progredire rapidamente. La disfunzione epatica progressiva nella EPP è associata con l'aumento della concentrazione di protoporfirina nel fegato, nel plasma e negli eritrociti, e con il peggioramento della fotosensibilità. Il danno epatico può essere in parte dovuto agli effetti tossici e colestatici dei grandi quantitativi di protoporfirina presenti nel fegato. Se è presente anche una patologia epatica concomitante, l'insufficienza epatica può essere reversibile. Altrimenti, il trattamento è generalmente inefficace e può divenire necessario il trapianto di fegato. Nelle forme avanzate dell'epatopatia da protoporfiria sono state osservate lesioni gravi da fotosensibilità simili a ustioni (specialmente dopo esposizione alle luci delle sale operatorie) e perfino una neuropatia motoria.
Diagnosi La EPP deve essere sospettata nei pazienti che lamentano fotosensibilità cutanea iniziata precocemente nel corso della vita, ma che non riferiscono la comparsa di vesciche o cicatrici. Un'anamnesi familiare negativa è di riscontro comune. La concentrazione di protoporfirina negli eritrociti e nel plasma è marcatamente aumentata, mentre le porfirine urinarie non lo sono. L'aumento della protoporfirina eritrocitaria non è specifico; esso può comparire nei deficit di ferro, nell'avvelenamento da piombo, in diversi disordini degli eritrociti, in ogni porfiria autosomica recessiva e talvolta nelle porfirie autosomiche dominanti acute. Un aumento dei livelli plasmatici delle porfirine, tuttavia, si verifica raramente in condizioni diverse dalle porfirie che provocano fotosensibilità cutanea. In tutte le condizioni diverse dalla EPP nelle quali la protoporfirina eritrocitaria è aumentata, comprese alcune altre porfirie, la protoporfirina in eccesso presente nei globuli rossi è complessata con lo zinco, mentre nella EPP essa non lo è. Le protoporfirine eritrocitarie complessate con lo zinco e non complessate con esso non vengono dosate separatamente nella maggior parte dei laboratori. Poiché si ritiene talvolta (non correttamente) che l'avvelenamento da piombo sia associato con un aumento della protoporfirina libera eritrocitaria, può risultare poco chiaro se il valore di una concentrazione di protoporfirina libera eritrocitaria si riferisca a quella complessata con lo zinco o a quella non legata al metallo. Nella EPP possono verificarsi aumenti notevoli della protoporfirina nelle feci. Il dosaggio delle porfirine negli eritrociti, nel plasma e nelle feci può essere utile per identificare la malattia nei familiari del paziente. La ricerca di una mutazione ereditaria della ferrochelatasi nei familiari è realizzabile se è stata identificata la mutazione esatta nel caso indice.
Terapia e prognosi La fotosensibilità viene trattata evitando l'esposizione alla luce solare. Il beta carotene, quando assunto in quantità tali da causare un lieve ingiallimento della cute, è particolarmente efficace per il trattamento di questa porfiria; 120-180 mg/ die PO migliorano la tolleranza alla luce solare in molti pazienti. Il livello sierico terapeutico raccomandato di beta carotene va da 600 a 800 mg/dl; gli effetti positivi si manifestano solitamente da 1 a 3 mesi dopo l'inizio della terapia. La protoporfirina escreta nella bile può essere parzialmente riassorbita dall'intestino e ritornare per via ematica al fegato. Al fine di interrompere questo circolo enteroepatico sono stati somministrati, con qualche successo, resine e altri agenti leganti. I farmaci dannosi per le porfirie epatiche non sembrano esacerbare le porfirie eritropoietiche, ma vengono comunque evitati per precauzione. Il deficit di ferro può contribuire alla compromissione dell'attività della ferrochelatasi e deve essere trattato adeguatamente. Diversamente dalle porfirie epatiche, il decorso della EPP è costante nel tempo e si osservano scarse variazioni dei livelli di protoporfirina nel plasma e negli eritrociti, salvo quando si sviluppano complicanze epatiche. Il trattamento dell'insufficienza epatica da protoporfirina è complesso e può richiedere il trapianto di fegato.
Le porfirie sono un gruppo di malattie metaboliche rare, per lo più ereditarie, causate dalla ridotta attività di uno degli enzimi della via biosintetica dell'eme (complesso ferroso costituente dell'emoglobina e di altre sostanze dell'organismo). Nelle varie forme, in base allo specifico difetto enzimatico, si determina iperproduzione e accumulo di varie sostanze dette porfirine (o dei loro precursori nella via metabolica) in sedi diverse, con diversa via di eliminazione e sintomatologia. La classificazione più usata è basata sui sintomi e distingue anzitutto tra porfirienon acute e acute. Tra le prime, di sintomatologia esclusivamente cutanea, la porfiria eritropoieticacongenita (morbo di Gunther) comporta anemia emolitica e grave fotosensibilità, presenti dalla nascita a causa della gran quantità di porfirine circolanti (un classico indizio è il pannolino del neonato rosso scuro per la gran quantità di porfirine eliminate con le urine). Caratteristica è anche l'eritrodonzia (pigmentazione rossa dei denti), dovuta alle porfirine che si depositano nel fosfato di calcio dentale. La porfiria cutanea tarda compare invece generalmente nella terza o quarta decade di vita, ed è rara nell'infanzia; la sintomatologia interessa le zone fotoesposte e comprende fragilità cutanea, bolle ed erosioni che finiscono in croste e talvolta in milii (piccole cisti), ipertricosi temporo-malare, iperpigmentazione, senescenza cutanea precoce e, talora, aspetti sclerodermiformi. Spesso si associa un'epatopatia di diverso grado ed eziologia, che può essere dovuta all'eccessiva assunzione di alcol, al sovraccarico di ferro, all'infezione da virus e qualche volta a farmaci, soprattutto estrogeni. La protoporfiria eritropoietica compare nei primi anni di vita e comporta bruciore, eritema ed edema delle zone fotoesposte e, solo raramente, bolle. Gli esiti della sintomatologia acuta sono caratterizzati da ispessimento della cute con aspetto di "acciottolato romano", soprattutto al dorso delle mani e al naso. Spesso vi è anche compromissione epatica, per la stasi di protoporfirine negli epatociti e nei canalicoli biliari, con conseguente formazione di calcoli nella colecisti. Le porfirie acute (porfiria acuta intermittente, deficit di ALA-deidratasi, coproporfiria ereditaria, porfiria variegata) hanno tutte sintomatologia neurologica. Nella porfiria variegata e nella coproporfiria ereditaria può esservi anche interessamento cutaneo. Tra i sintomi più frequenti presenti nelle diverse forme di porfirie acute - con particolare riferimento alla porfiria acuta intermittente e al deficit di ALA-deidrasi - vi sono attacchi ricorrenti di dolori addominali e disturbi gastroenterici (nausea, vomito, stipsi), oltre ad una serie di problemi neurologici di vario livello. Per la diagnosi sono necessari l'esame clinico e la valutazione delle analisi di laboratorio, tra cui la determinazione qualitativa e quantitativa delle porfirine nei diversi materiali biologici e quella dei precursori delle porfirine nelle urine; va poi determinata anche l'attività enzimatica, che consente di individuare i portatori asintomatici. Oggi, dopo l'individuazione dei cromosomi che ospitano i geni dei diversi enzimi coinvolti nella biosintesi dell'eme, prosegue la definizione delle specifiche mutazioni. Date le differenze biochimiche e cliniche tra le varie forme, bisogna adottare schemi terapeutici differenziati (anche farmacologici). Oggi esistono sostanze che danno dei risultati nel trattamento delle porfirie, mirando ai seguenti obiettivi: prevenire i fattori scatenanti, diminuire la produzione di porfirine e di precursori, favorirne l'eliminazione, agire sui sintomi.