Download

1 / 53
580 likes | 689 Views
Amplifying DNA: PCR & cell-based DNA cloning. The importance of DNA cloning: Current DNA technology is based on two different approaches:

E N D
Amplifying DNA: PCR & cell-based DNA cloning • The importance of DNA cloning: • Current DNA technology is based on two different approaches: a. Specific amplification (DNA cloning) which involves cell-based DNA cloning (involving a vector/replicon and a suitable host cell) andin vitro DNA cloning (PCR) b. Molecular hybridization where the DNA fragment of interest is specifically detected using a mixture of different sequences
2. Polymerase Chain Reaction (PCR): features & Applications: • Template DNA: DNA (linear or circular) or cDNA (complementary DNA produced from produced mRNA by reverse transcriptase) • Primers: pairs of oligonucleotides each 18-25 nucleotides long; 40%-60% GC content; melting temp of both should not differ by >5oC; 3’ terminal sequences of any primer should not be to any sequences of the other primer in the pair; self-complimentary sequences (inverted repeats) of >3 bp should be avoided. • Cycling nature & exponential amplification: denaturation; primer annealing; and DNA synthesis (extension). • Regular Taq DNA polymerase lacks 3’ -> 5’ exonuclease activity needed to provide proof-reading function.
PCR has two limitations: a.short sizes of amplified products (<5 kb). This is solved by doing Long-range PCR (up to tens of Kb long) which uses a mixture of two heat stable polymerases that provide optimal levels of DNA synthesis as well as a 3’ -> 5’ exonuclease activity. b.low yields of amplifications which is resolved by cloning the PCR amplified DNA fragment in a vector then propagating the vector in a cell based system (clone by A/T cloning or by using anchored PCR primers).
General Applications of PCR: PCR has 3 major advantages: - rapid - sensitive - robust (possible to amplify DNA from damaged tissues or degraded DNA)
Primer specificity is very important in PCR. Several modifications have been developed to reduce nonspecific binding (see Box 5.1): - Hot-start PCR - Nested PCR - Touch-down PCR • The correct base pairing at the extreme 3’ end of bound primers is a requirement for producing a PCR product. This allowed the use of PCR to distinguish between alleles of the same gene that differ in a single nucleotide (allele-specific PCR). This method is known as ARMS (amplification refractory mutation system).
Degenerate oligonucleotide primed PCR (DOP-PCR) allow the amplification of a different but closely related genes (novel genes) at the same time. • Indiscriminate amplification of whole genomes can be performed using linker-primed PCR (ligation adaptor PCR). • PCR could be used to amplify unknown DNA sequences neighboring a known sequence. Such methods include anchored PCR, inverse PCR, RACE-PCR.
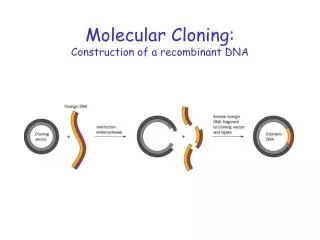
Principles of cell-based cloning: • Four steps in cell-based cloning - Construction of recombinant DNA molecules. Involves the use of endonuclease restriction enzymes, ligation, and a replicon (vector). - Transformation in appropriate host cells. - Selective propagation of cell clones. This step takes advantage of selectable markers. - Isolation of recombinant DNA from cell clones followed by molecular characterization (such as restriction enzyme analysis).
Endonuclease restriction enzymes type II (RE), are a powerful tool (molecular scissors) used in restricting target DNA (whole genome or plasmid) into smaller DNA fragments. The restriction of a DNA double helix molecule may result in a blunt end or a cohesive end terminus (sticky end generating a 3’ or 5’ single strand overhang). See Table 5.1 • RE are used to generate recombinant linear molecules (concatemers) or circular molecules (cyclization). • Simple cloning vectors include bacterial plasmids and bacteriophages.
Recombinant DNA molecules are transferred into appropriate host cells (e.g. bacteria) for propagation. Normally a single recombinant DNA exists per cell but sometimes co-transformation may result in two or more recombinant DNA molecules per host cell.
DNA libraries are a collection of clones that represent the entire genome of an organism. Two types of libraries are known: - Genomic library. To be representative of the entire genome, the library should be >4GE. A genome equivalent (GE) is genome size/average insert size. In humans, for a GE=1, you need 3000Mb genome size)/40 kb (insert size) = 75,000 independent clones. - cDNA library. Takes advantage of reverse transcriptase. Usually much smaller than a genomic library.
Selection of recombinant clones necessitates the use of an appropriate selectable marker system. - screening by vector molecules which includes antibiotic resistance genes or β-galactosidase gene complementation - Generalized recombinant screening by insertional inactivation. This can be achieved by β-galactosidase screens or suppressor t-RNA-based screens. - Directed recombinant screening. This can be achieved by hybridization-based screening by using labeled probes or by using PCR-based screening.
4. Cloning systems for different sized DNA fragments: • Such cloning systems normally include an antibiotic resistance gene (to enable screening for presence of vector) and a marker gene with a multiple cloning site (to enable screening recombinant clones). • See Table 5.2 for different cloning vectors and the DNA insert sizes that each could accommodate.
Lambda and cosmid vectors are used in cloning moderately large DNA fragments in bacterial cells. Three types of λ derived cloning vectors: a. Replacement λ vectors: removal of central section of the genome and replacing by a foreign DNA fragment (up to 23 kb inserts) b. Insertion λ vectors: modifications to allow insertional cloning of cDNA fragments into the cI gene (up to 5 kb) c. Cosmid vectors: cos sequences of λ are inserted into a small plasmid generating a cosmid. Can take 33 – 44 kb inserts.
Bacterial Artificial Chromosome (BAC) vectors are used to clone large fragments (>300 kb). Low copy number (1-2 copies/cell) fertility factor (F-factor) plasmids are used for this purpose. • Bacteriophage P1 vectors and P1 artificial chromosomes (PACs). Components of the P1 phage are included in a circular plasmid and can accommodate up to 122 Kb DNA fragments.
Yeast Artificial Chromosomes (YACs) permit the cloning of 0.2 – 2.0 Megabases. YACs are propagated in yeast as a linear chromosome which becomes part of the genome and is distributed by the mitotic machinery. YACs must include: - centromere sequences (CEN) - Telomere sequences (TEL) - Autonomous replicating sequences (ARS) for replication in the yeast nucleus. - Ampicillin resistance for propagation in E. coli - Three markers including a suppressor tRNA gene, TRP1, and URA3 genes for selection by complementation in the appropriate yeast host cell.
5. Cloning systems for producing mutagenized DNA: • Cell-based oligonucleotide mismatch mutagenesis can be used to generate a specific nucleotide substitution in a coding sequence of a gene. This is achieved by using M13 vectors to generate single-stranded recombinant DNA
Production of single-stranded DNA for use in sequencing is obtained using M13 vectors or phagemid vectors.
PCR-based mutagenesis could be used to achieve two types of changes: - 5’ adds-on mutagenesis which adds specific sequences at the 5’ of the amplified product. Such sequences may include a phage promoter to drive gene expression. - Site-directed mutagenesis which results in an amplified product with a specific base substitution to introduce a specific amino acid substitution at the protein level.
Cloning systems designed for gene expression: • Bacterial cells are used as hosts for recombinant expression vectors designed for the production of large amounts of a recombinant protein (fusion proteins or tagged proteins). • Problems with overexpression in bacteria include toxicity of large amounts of the recombinant protein, lack of posttranslational processing, inability to synthesize very large mammalian proteins, and protein folding and solubility.
To solve the above mentioned problems: - The use of pET-3 bacterial vectors containing T7 promoter in combination with host cells carrying the gene for T7 RNA polymersae expressed under the control of the lacZ promoter (i.e. inducible by IPTG). - The vector is designed so that the recombinant protein is fused to an endogenous protein (fusion proteins). - Use an affinity tag so that the recombinant fusion protein be purified by affinity chromatography. Two affinity tagging systems are GST-glutathione affinity and polyhistidine-nickel ion affinity.