Download
1 / 20
200 likes | 325 Views
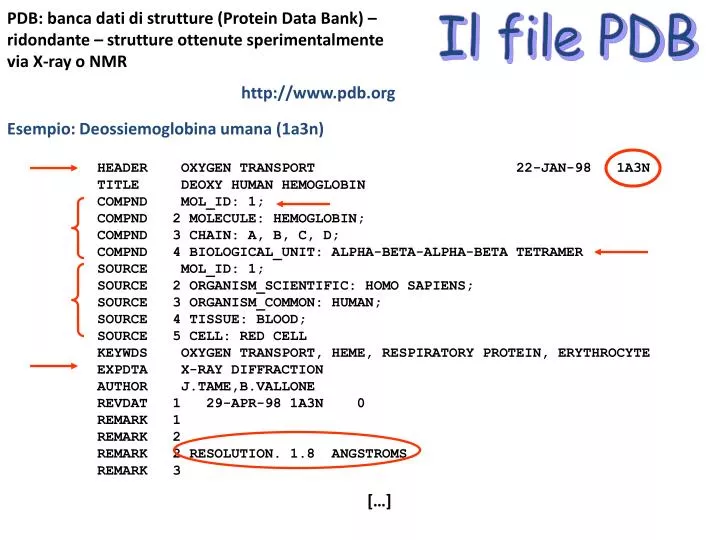
PDB: banca dati di strutture (Protein Data Bank) – ridondante – strutture ottenute sperimentalmente via X-ray o NMR. Il file PDB. http://www.pdb.org. Esempio: Deossiemoglobina umana (1a3n).

E N D
PDB: banca dati di strutture (Protein Data Bank) – ridondante – strutture ottenute sperimentalmente via X-ray o NMR Il file PDB http://www.pdb.org Esempio: Deossiemoglobina umana (1a3n) HEADER OXYGEN TRANSPORT 22-JAN-98 1A3N TITLE DEOXY HUMAN HEMOGLOBIN COMPND MOL_ID: 1; COMPND 2 MOLECULE: HEMOGLOBIN; COMPND 3 CHAIN: A, B, C, D; COMPND 4 BIOLOGICAL_UNIT: ALPHA-BETA-ALPHA-BETA TETRAMER SOURCE MOL_ID: 1; SOURCE 2 ORGANISM_SCIENTIFIC: HOMO SAPIENS; SOURCE 3 ORGANISM_COMMON: HUMAN; SOURCE 4 TISSUE: BLOOD; SOURCE 5 CELL: RED CELL KEYWDS OXYGEN TRANSPORT, HEME, RESPIRATORY PROTEIN, ERYTHROCYTE EXPDTA X-RAY DIFFRACTION AUTHOR J.TAME,B.VALLONE REVDAT 1 29-APR-98 1A3N 0 REMARK 1 REMARK 2 REMARK 2 RESOLUTION. 1.8 ANGSTROMS. REMARK 3 […]
tipo di atomo tipo di amminoacido coordinate X Y Z ATOM 1 N VAL A 1 10.720 19.523 6.163 1.00 21.36 N ATOM 2 CA VAL A 1 10.228 20.761 6.807 1.00 24.26 C ATOM 3 C VAL A 1 8.705 20.714 6.878 1.00 18.62 C ATOM 4 O VAL A 1 8.164 20.005 6.015 1.00 19.87 O ATOM 5 CB VAL A 1 10.602 22.000 5.966 1.00 27.19 C ATOM 6 CG1 VAL A 1 10.307 23.296 6.700 1.00 31.86 C ATOM 7 CG2 VAL A 1 12.065 21.951 5.544 1.00 31.74 C ATOM 8 N LEU A 2 8.091 21.453 7.775 1.00 16.19 N ATOM 9 CA LEU A 2 6.624 21.451 7.763 1.00 17.31 C ATOM 10 C LEU A 2 6.176 22.578 6.821 1.00 18.55 C ATOM 11 O LEU A 2 6.567 23.730 7.022 1.00 18.72 O ATOM 12 CB LEU A 2 6.020 21.707 9.129 1.00 18.34 C ATOM 13 CG LEU A 2 6.386 20.649 10.198 1.00 17.39 C ATOM 14 CD1 LEU A 2 5.998 21.119 11.577 1.00 17.99 C ATOM 15 CD2 LEU A 2 5.730 19.337 9.795 1.00 16.96 C ATOM 16 N SER A 3 5.380 22.237 5.852 1.00 15.02 N ATOM 17 CA SER A 3 4.831 23.237 4.928 1.00 16.59 C ATOM 18 C SER A 3 3.725 24.027 5.568 1.00 14.84 C ATOM 19 O SER A 3 3.095 23.717 6.591 1.00 14.40 O ATOM 20 CB SER A 3 4.308 22.429 3.727 1.00 16.47 C ATOM 21 OG SER A 3 3.076 21.786 3.991 1.00 14.91 O …
SCOP (StructuralClassificationof Proteins) • Class • a, b, a/b, a+b, ... • Fold • Similarità strutturale • Superfamily • Omologia • Family • Omologia e funzione • Principalmente annotata a mano
CATH • Class (a, b, a/b, a+b, ...) • Architecture • Topology (including connections between sec. str. elements) • Homologous superfamily • Semiautomatica • Solo Architecture viene assegnata manualmente
CONFRONTO STRUTTURE 3D DI PROTEINE – ALLINEAMENTO STRUTTURALE Come nel confronto di sequenze e’ necessario allinearle, nel confronto di strutture 3D e’ necessario sovrapporle come corpi rigidi scegliendo una regola di corrispondenza tra coppie di atomi o di residui nelle due strutture. La prima difficoltà consiste nel fatto che le due proteine molto spesso non hanno lo stesso numero di residui. Per la sovrapposizione si possono utilizzare le catene dei carboni alfa appartenenti agli elementi di struttura secondaria perche’ in genere le inserzioni e delezioni si accumulano nei loop che possono semplicemente venire esclusi dalla sovrapposizione. I metodi di confronto 3D utilizzano l’ allineamento delle sequenze per decidere la regola di corrispondenza alla base della sovrapposizione strutturale
Un allineamento strutturale può essere valutato in base alla deviazione quadratica media (root mean square deviation o r.m.s.d.), al numero di atomi che sono stati accoppiati nella sovrapposizione e alla valutazione della similarità dei residui sovrapposti. D = distanza tra coppie di atomi appaiati N = numero di coppie considerate L’r.m.s.d. o r.m.s. di una sovrapposizione tridimensionale è la distanza media tra gli atomi di tutte le coppie che hanno partecipato all’allineamento strutturale, per cui tanto più bassa è l’r.m.s. tanto migliore sarà l’allineamento strutturale calcolato
Root-mean-square deviation Deviazione quadratica media Serve per paragonare strutture identiche, eccetto rotazioni e traslazioni rai e rbi sono le posizioni dell´ atomo i nelle strutture a e b, n è il numero di atomi nelle strutture. RMSD r.m.s.d. di una sovrapposizione 3D è la distanza media tra gli atomi di tutte le coppie che hanno partecipato all’allineamento strutturale, per cui tanto più bassa è l’r.m.s. tanto migliore sarà l’allineamento strutturale calcolato
EVOLUZIONE DELLE PROTEINE RELAZIONE ESISTENTE TRA SIMILARITA’ di sequenza e struttura in omologhi Studio di un campione di strutture note di proteine omologhe (Chothia, Lesk, 1982) Similarità strutturale misurata in termini di RMSD CALCOLO RMS • Confronto di due strutture mediante SOVRAPPOSIZIONE • Determinazione di aa che si corrispondono nelle due strutture • Identificazione degli atomi che si vogliono confrontare (Cα o backbone) • RMSD= n = numero residui d = distanza tra atomi corrispondenti
valutazione dell’allineamento strutturale un altro criterio di valutazione di un allineamento strutturale è rappresentato dal numero di atomi o di residui che sono stati accoppiati si cerca di massimizzare il numero di atomi accoppiati e di minimizzare la corrispondente r.m.s. a parità di numero di residui accoppiati, il migliore allineamento strutturale sarà quello con minore r.m.s. a parità di r.m.s. verrà considerato migliore l’allineamento strutturale operato con un maggior numero di atomi accoppiati oltre a questi due valori tipici delle sovrapposizioni tridimensionali, si può anche considerare il punteggio di similarità dei residui accoppiati
DIVERGENZA DI SEQUENZA E STRUTTURA 30% id.seq RMSD (A) Se si valuta la relazione esistente tra divergenza strutturale (misurata in termini di RMSD) e divergenza di sequenza si osserva che esse si corrispondono in MODO ESPONENZIALE (valutato dal confronto di strutture note) %identity • (RMSD< 2 Å) possono avere seq molto differenti • (degenerazione del codice strutturale-struttura più conservata della seq)
SOGLIA di significatività per la SIMILARITA’ STRUTTURALE 100 Sec str identity < 70% %id seq Sec str identity > 70% 30 .confronto a coppie di struttura e seq di proteine 3D note, quantificato usando come parametri: lunghezza allineamento, % idseq, sim.struttura a confronto in un grafico: lunghezza allin vs idseq (e in terza dimensione sim.strutturale) Sim. Strut.parametrizzata come uguale/diverso (pallino/quadrato): 2 str. Uguali se > 70% della struttura sec è in comune (rmsd bassa). al di sotto della soglia rumore di fondo elevato 25% (x allin 80 aa) al di sopra del quale chiara similarità strutturale – soglia lunghezza dipendente possibile che proteine con identità < 25% abbiano strutture simili ma anche che non siano correlate strutturalmente (twilight zone) 0 0 Length alignment 150
Proteinubiquitination Proteinubiquitination: regulatoryprocessinfluencingseveralaspectofeukaryoyiccellbiology Polyubiquitin (polyUb) chain: link ε-amino groupof a LysofoneUbto the C-terminalcarboxylgroupof the nextUb in the chain E1-E2-E3 responsibleforactivating and transferringUbtoproteins FundamentalmechanismsofUb-chainassembly and of E1-E2-E3 regulationstillnotclearlyunderstood • Passmore, and Bardford, Biochem J, 2004 • Pickart, Cell, 2004 • Brzovic and Klevit, Cell Cycle, 2006 • Hochstrasser, Cell, 2006
Ubiquitinlikeproteins(Ubls) 8-20 kDa Ub 76 aa Globular domain (4βstrands in a antiparallelβsheet +αhelix) + flexibleC-terminalterminatingwith a gly 7 Conservedlysineresidues in Ub N.B. ISG15: ha due domini Ubl Di-Ubiquitina Ogni Ubl ha il suo set di E1-E2-E3
E2: Ub-conjugatingenzymes Cysforms a thiolester bond toC-terminalGlyofUb Asnstabilizesoxyaniontransition state E1 E3/E3 Multi-domain E2 classification in 17 familesreflectingstructuralorganizationof cat. dom. IdentificationofphosphorylationsbyCdks and CK2 kinasesaffecting E2 activity • Michelle et al., J Mol Evol, (2009) • Ye and Rape, Nat Rev Mol Cel Biol (2009)
MonomericUb K48 Hydrophobic patch G76 K63
B_Cterm K48-linked polyUb Diverse strutture sperimentali di Di-Ubcrosslinkata K48 via NMR o X-ray e una struttura X-ray della tetra-polyUbcrosslinkata via K48 B_VAL70 B_LEU8 A_ILE44 B_ILE44 A_LEU8 A_VAL70 Legame isopeptidico polyUb crossilinkata by K48 sono segnale universale per la degradazione da proteasoma Adottano una CONFORMAZIONE CHIUSA in cui L8, I44 and V70 (patch idrofobico sulla superficie di una singola molecola di Ub) sono sequestrati all’interfaccia tra due molecole di Ub adiacenti
K63-linked polyUb Diverse strutture sperimentali di Di-Ubcrosslinkata K63 via NMR o X-ray e una struttura X-ray della tetra-polyUbcrosslinkata via K63 polyUbcrosslinkata via K63 agisce come segnale regolatorio piuttosto che degradativo in molti pathway, Adotta conformazione estesa in soluzione senza contatti diretti tra patch idrofobici






















