Download

1 / 53
630 likes | 769 Views
This guide covers the workflow for exome sequencing data analysis including read generation, mapping, variant calling, annotation, and filtering. Learn about driver mutations and tools like BWA, SAM/BAM, GATK, and more. Enhance your understanding of SNP calling, short indels, structural variations, and filtering techniques for driver mutations. Improve your data analysis skills in exome sequencing to detect differences to the reference genome. Examples and useful links provided in the text.

E N D
Data Analysis for Exome Sequencing Data Chih-Hao Hsu 03/18/2015
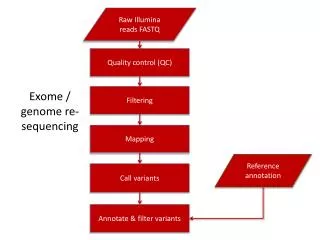
Workflow for Data Analysis Read Generation Read Mapping Variant Calling Annotation and Filtering Driver Mutations
Workflow for Data Analysis Read Generation - Store in a FASTQ file - QC study Read Mapping Variant Calling Annotation and Filtering Driver Mutations
Raw Sequence Data Format • FASTQ format • Phred quality score Sequence ID Sequence Quality score !"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRS Phred score 0………………………………………………………………………………………….50 Error rate 1……………………………………………………………………………………..0.00001 Phred score = -10 * log10P
Sequence quality: FastQC • http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Workflow for Data Analysis Read Generation - Map reads to reference genome - Different aligners - SAM/BAM file - BAM improvement Read Mapping Variant Calling Annotation and Filtering Driver Mutations
Read Mapping • Challenge: • compare billion of short sequence reads against human genome (3Gb) • Burrows-Wheeler Alignment tool (BWA) • Popular tool for genomic sequence data • “index” the human genome to allow memory-efficient and fast string matching between sequence read and reference genome
Different Alignment Algorithms • BWA – 2009 • BWA-SW – 2010 • BWA-MEM – 2013 • Bowtie – 2009 • Bowtie2 – 2012 • Gem – 2012 • Cushaw2 – 2014 • Novoalign Li, arXiv:1303.3997 (2013)
SAM/BAM Format • SAM (Sequence Alignment/Map) format • Single unified format for storing read alignments to a reference genome • BAM (Binary Alignment/Map) format • Binary equivalent of SAM • Advantages • Supports indexing • Compact size
1000 Genomes BAM File Header Data
BAM Visualization Mismatches Reference
BAM Improvement • Remove duplicates • Local realignment • Base quality recalibration
Library Duplicates • All second-gen sequencing platforms are NOT single molecule sequencing • PCR amplification step in library preparation • Can result in duplicate DNA fragments in the final library prep. • PCR-free protocols do exist – require large volumes of input DNA • Can result in false SNP calls • Duplicates manifest themselves as high read depth support
Remove Duplicates • Identify read-pairs where the outer ends map to the same position on the genome and remove all but 1 copy • Samtools: samtools rmdup or samtools rmdupse • Picard/GATK: MarkDuplicates
Local Realignment - indels • The trouble with mapping approaches
Local Realignment - indels • The trouble with mapping approaches
Local Realignment - indels • The trouble with mapping approaches
Local Realignment - indels Local realignment
Local realignment in GATK • Uses information from known SNPs/indels (dbSNP, 1000 Genomes) • Uses information from other reads • Smith-Waterman exhaustive alignment on select reads
The quality scores issued by sequencers are inaccurate and biased • Quality scores are critical for all downstream analysis • Systematic biases are a major contributor to bad calls https://www.broadinstitute.org/gatk/
Base Quality Recalibration in GATK • Align subsample of reads from a lane to human reference • Exclude all known dbSNP sites • Assume all other mismatches are sequencing errors • Compute a new calibration table based on mismatch rates per position on the read
Workflow for Data Analysis Read Generation Read Mapping Variant Calling - SNP calling - Short Indels - Structural Variation - Germline vs. Somatic - VCF files Annotation and Filtering Driver Mutations
Variant Calling Differences to the reference Reference: C Sample: T
Signal vs. Noise Sanger: is it real?? Total count: 204 A : 18 (9%, 12+, 6-) C : 1 (0%, 0+, 1-) G : 0 T : 185 (91%, 92+, 93-) N : 0 NGS: read count Provides confidence (statistics!) Sensitivity tune-able parameter (dependent on coverage)
Variant Calling • SNP calling • Short Indels • Structural Variation
SNP Calling • SNP – single nucleotide polymorphisms • Examine the bases aligned to position and look for differences • Factors to consider when calling SNPs • Base call qualities of each supporting base • Proximity to small indel • Mapping qualities of the reads supporting the SNP • Read length • Paired reads • Sequencing depth
Example SNP http://www.sanger.ac.uk/mousegenomes
Is this a SNP? http://www.sanger.ac.uk/mousegenomes
Short indel Calling • Small insertions and deletions observed in the alignment of the read relative to the reference genome • Factors to consider when calling indels • Misalignment of the read • Homopolymer runs either side of the indel • AAAA or TTTTTTTT • Length of the reads
Example Indel http://www.sanger.ac.uk/mousegenomes
Is this a Indel? http://www.sanger.ac.uk/mousegenomes
Germline vs. Somatic Variants • Genes and chromosomes can mutate in either somatic or germline tissue Mutation Detection
An Example of Germline Variants Robinson et al. 2011
An Example of Somatic Variants Normal Tumor
Different Variants Callers Ding, Nat Rev Genet. 2014
The GATK software • Genome Analysis Toolkit, BROAD Institute http://www.broadinstitute.org/gatk/ • Initially developed for 1000 Genomes Project • Single or multiple sample analysis (cohort) • Popular tool for germline variant calling
Somatic Variant Calling • Somatic mutations can occur at low freq. (<10%) due to: • Tumor heterogeneity (multiple clones) • Low tumor purity (% normal cells in tumor sample) • Requires different thresholds than germline variant calling when evaluating signal vs noise • Trade-off between sensitivity (ability to detect mutation) and specificity (rate of false positives)
ICGC-TCGA DREAM Mutation Calling challenge • MuTect ranked highly in all 4 datasets in the DREAM challenges
Variant Call Format (VCF) • VCF is a standardized format for storing DNA polymorphism data • SNPs, insertions, deletions and structural variants • With rich annotations • Indexed for fast data retrieval of variants from a range of positions • Store variant information across many samples • Record meta-data about the site • dbSNP accession, filter status, validation status, • Very flexible format
Workflow for Data Analysis Read Generation Read Mapping Variant Calling Annotation and Filtering - Genome Annotation Database - Criteria for filtering Driver Mutations
dbSNP • dbSNP is a free public archive for genetic variation within and across different species developed by NCBI Sherry, Genome Res. 1999
1000 Genomes Project • 15 million SNPs • 1 million short insertions/deletions • 20,000 structural variants The 1000 Genomes Project Consortium, Nature 2010 (www.1000genomes.org)
COSMIC • COSMIC is the most comprehensive resource for exploring impact of somatic mutations in human cancer Forbes, Nucleic Acids Research 2015
COSMIC http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/
Identifying causal variants: filtering Stitziel, Genom Biol 2011
Workflow for Data Analysis Read Generation Read Mapping Variant Calling Annotation and Filtering - Significantly mutated genes - Pathway and network analysis Driver Mutations