Download

1 / 24
670 likes | 1.52k Views
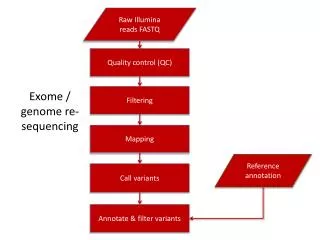
Whole Exome Sequencing for Variant Discovery and Prioritisation. Exomes: Publication Trends. Total: 925 (Oct 2012). 2013: ~ 800 papers 2014: ~ 1200 papers. Forero DA, 2012. NGS Variation Discovery Workflow ( resequencing based). Variant Discovery Application: Disease.

E N D
Whole Exome Sequencing for Variant Discovery and Prioritisation
Exomes: Publication Trends Total: 925 (Oct 2012) 2013: ~ 800 papers 2014: ~ 1200 papers Forero DA, 2012
Variant Discovery Application: Disease • An equivalent of the genome would amount almost 2000 books, containing 1.5 million letters each (average books with 200 pages)! • This information is contained in any single cell of the body.
Monogenic Diseases • Single mutation • How do we find it in all those ‘books’? • A bioinformatics challenge • NGS sequencers can only read small portions • So, the library is fragments of pages of the books!
Mendelian Disease Gene Discovery Gilissen, Genome Biol 2011
Mendelian Disease Gene Discovery Gilissen, Genome Biol 2011
Opportunities and Challenges • Exomes more cost effective: Sequence patient DNA and filter common SNPs; compare parents child trios; compare paired normal cancer • Challenges: • Still can’t interpret many Mendelian disorders • Rare variants need large samples sizes • Exome might miss region (e.g. novel non-coding genes) Shendure, Genome Biol 2011
Why exome sequencing? • WGS still too costly • WES: targets ~1% of human genome) • Mendelian disordersmostly disrupts protein-coding sequences • Large fraction of rare non-synonymous variants in human genome are predicted to be deleterious • Splice sites also enriched for highly functional variation • Search for variants with large effect sizes
A representation of the relationship between the size of the mutational target and the frequency of disease for disorders caused by de novo mutations Gilissen, GenomBiol 2011
Maximizing chances of finding disease-causing rare variants using exome sequencing Bamshad, Nat Rev Genet 2011
Example: Comparative Sequencing • Somatic mutation detection between normal / cancer pairs • More mutation yield and better causal gene identification than Mendelian disorders Meyerson et al, Nat Rev Genet 2010
BUT Exome Analysis for a single patientcan be informative Perraultsyndrome (HSD17B4) Pierce, Am J Hum Genet 2010
Read Mapping • Mapping hundreds of millions of reads the reference genome is CPU and RAM intensive, and ‘slow’ • Read quality decreases with length (small single nucleotide mismatches or indels – real or artifact?) • Very few mappers appropriately deal with indels • Mapping output: SAM (BAM) or BED
Mapped Data: SAM specification • Simple generic sequence alignment format • Describes alignment of reads to a reference • Flexible - stores all the alignment information • Keeps track of chromosome position, alignment quality and alignment features (extended cigar) • Includes mate pair / paired end information • Original FASTQ data can be reproduced from SAM (and BAM)
BAM format • Binary version of SAM - more compact • Makes downstream analysis independent from the mapping program • Allows most of operations on alignment to work on a stream without loading the whole alignment into memory • Allows the file to be indexed by genomic position to efficiently retrieve all reads aligning to a locus
VCF format • Emerging standard for storing variantdata • Originally designed for SNPs and short INDELs, it also works for structural variations • Consists of header and data sections • The data section is TAB delimited with each line consisting of at least 8 mandatory fields
Variant Prioritization • Heuristic filtering to identify novel genes for Mendelian disorders Stitziel et al, Genome Biol 2011
Example WES-based variant discovery workflow • Map the reads to a reference genome • index the reference genome • Map (BWA, BOWTIE, NOVOAOLIGN, ETC) • Sort BAM file • Remove PCR duplicates • Realign around indels (‘optional’) • Call variants • Recalibrate quality scores (‘optional’) • Filter variants • Basic variant annotation • Biological interpretation only starts here