Download
1 / 1
10 likes | 66 Views
Explore phase transition & self-assembly of lower diamandoids & derivatives via DFT-optimized MD simulations. Investigate bonding types & density effects on transition points.

E N D
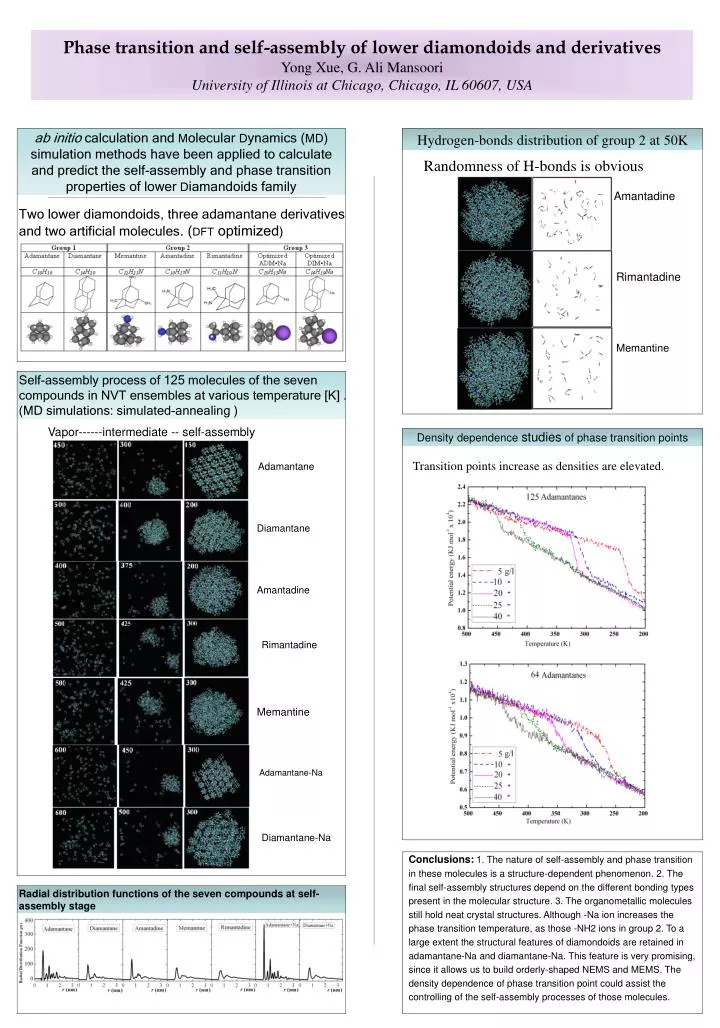
ab initio calculation and Molecular Dynamics (MD) simulation methods have been applied to calculate and predict the self-assembly and phase transition properties of lower Diamandoids family Hydrogen-bonds distribution of group2at50K Randomness of H-bonds is obvious Phase transition and self-assembly of lower diamondoids and derivativesYong Xue, G. Ali Mansoori University of Illinois at Chicago, Chicago, IL 60607, USA Amantadine Two lower diamondoids, three adamantane derivatives and two artificial molecules. (DFT optimized) Rimantadine Memantine Self-assembly process of 125 molecules of the seven compounds in NVT ensembles at various temperature [K] . (MD simulations: simulated-annealing ) Vapor------intermediate -- self-assembly Density dependence studies of phase transition points Transition points increase as densities are elevated. Adamantane Diamantane Amantadine Rimantadine Memantine Adamantane-Na Diamantane-Na • Conclusions:1. The nature of self-assembly and phase transition in these molecules is a structure-dependent phenomenon. 2. The final self-assembly structures depend on the different bonding types present in the molecular structure. 3. The organometallic molecules still hold neat crystal structures. Although -Na ion increases the phase transition temperature, as those -NH2 ions in group 2. To a large extent the structural features of diamondoids are retained in adamantane-Na and diamantane-Na. This feature is very promising, since it allows us to build orderly-shaped NEMS and MEMS. The density dependence of phase transition point could assist the controlling of the self-assembly processes of those molecules. Radial distribution functions of the seven compounds at self-assembly stage