Download

1 / 30
310 likes | 1.15k Views
Steroid/Intracellular Receptor Pharmacology. Therapeutic uses of agonist/antagonists Replacement therapy adrenal steroids for Addison’s disease estrogen and progesterone for menopause thyroid (hypothyroid disorders) Pharmacologic (non-physiological) uses

E N D
Steroid/Intracellular Receptor Pharmacology • Therapeutic uses of agonist/antagonists • Replacement therapy • adrenal steroids for Addison’s disease • estrogen and progesterone for menopause • thyroid (hypothyroid disorders) • Pharmacologic (non-physiological) uses • glucocorticoids as anti-inflammatory agents • estrogen/progesterone for contraception • androgens for increased muscle mass (abused by athletes) • mifepristone (RU486) for pregnancy termination • Cancer chemotherapy • tamoxifen, anastrozole for breast cancer • flutamide/bicalutamide, leuprolide for prostate cancer
AF2 AF1 a b LXR oxysterols FXR farnesoids PXR zenobiotics CAR phenobarbital Vitamin D, thyroid hormone, Retinoic Acid Receptors & Adopted Orphan Receptors Classical Steroid Hormone Receptors: these are a small subset of the 48 Nuclear/Intracellular Receptor Family members found in the human genome.
There are two genes coding for estrogen receptors in the human genome: ERa and ERb 18% 97% 60% homology (there appears to be no AF-1 function in human ERb) They bind the same DNA sequences, dimerize with themselves and each other. Are they just redundant or do they have specific physiological roles?
Expression patterns of ERs ERa breast uterus cervix vagina brain regions ERb ovary prostate, testis spleen, thymus lung hypothalamus, other brain regions Knockout phenotype: female sterility, no mammary gland development, obesity, male sterility, epididymal dysfunction, testicular degeneration Knockout phenotype: male--fertile, female--reduced fertility, ovarian dysfunction, vascular problems/develop hypertension as they age
antagonists or partial agonists agonists The classic view of estrogen action: E binds to ER (displaces a complex of chaperone proteins), ER forms dimers and interacts with the ERE to activate genes by recruitment of co-activators.
How does estrogen act in classical mode to induce gene expression from genes that have EREs in their promoter?
However, a number of observations do not fit this model some genes induced by E do not have a recognizable ERE tamoxifen can inhibit E induction of genes in breast but can be stimulatory of E responsive genes in uterus some of the effects of E are too fast to be transcriptional (e.g. rapid activation of the MAP kinase pathway in <5 min.) pharmacological actions of agonists and antagonists do not make sense in the classical model
new observation: ER does not need to bind to an ERE in order to induce gene expression. Induction can occur when ER binds indirectly to other transcription factors such AP1 and then recruits coactivators. classic pathway (fos/jun) indirect gene activation pathway
luciferase luciferase The regulation of ERE versus AP1-activated promoters depends on the receptor subtype, the ligand, and the cell type ERa ERb Promoter E TAM E TAM ERE AP1
A Structural basis for the differences in ER activity when agonist or antagonist ligands are bound the C-terminal trans- activation helix (H12) is in green AF2
Steroid Receptors also signal through non nuclear mechanisms to activate MAP kinase
Summary and prospects for the future 1. estrogen receptor ligands can act through multiple mechanisms: (1) classical ligand dependent induction thru an ERE (2) indirect ligand dependent induction by interaction with AP1 or perhaps other DNA bound transcription factors. (3) ligand-dependent interactions with Src kinase in the cytoplasm and the activation of Map kinase signaling 2. ligands can have agonist or antagonist properties depending on whether the receptor is ERa or ERb and the tissue complement of co-regulators 3. the complexity of biological responses can be utilized pharmacologically by the design of SERMs (selective estrogen receptor modulators --other nuclear receptors such as AR, GR may also be approached this way.
genistein: a phytoestrogen that is a selective estrogen receptor modulator (SERM): every morning with breakfast!
Results from the 2002 Women’s Health Initiative Study on Hormone Replacement Therapy in Postmenopausal Women
WHI subject profile • note: • many subjects had been E deficient for 10 yrs • high percentage were overweight. • Conjugated equine estrogen (a mixture of estrogens from pregnant horse urine) and medroxyprogesterone were given orally--the liver sees highest conc.
Corticosteroid Receptors AF2 AF1 a b LXR oxysterols FXR farnesoids PXR zenobiotics CAR phenobarbital cortisol binds to both GR and MR aldosterone specific for MR (mineralcorticoid receptor) Vitamin D, thyroid hormone, Retinoic Acid Receptors & Adopted Orphan Receptors
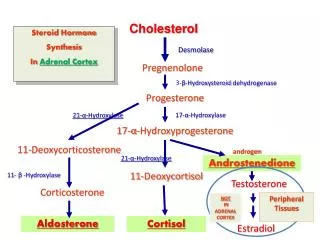
Therapeutic use of adrenal steroids • 1) Replacement therapy • Addison’s disease: administer a glucocorticoid (e.g. cortisol) • and a mineralocorticoid (e.g. fludrocortisone) Congenital adrenal hyperplasia • 21 b-hydroxylase deficiency is the most common cause • 2) Diagnosis of Cushing’s syndrome • 3) Cancer chemotherapy (especially lymphoma/leukemia) • 4) Anti-inflammatory agents
The HPA axis regulates the immune system, the musculoskeletal system and many tissues involved in overall metabolism like liver and fat.
Physiological effects of glucocorticoids Metabolic gluconeogenesis (liver) release of amino acids (muscle) release of fatty acids-lipolysis (fat) plasma glucose insulin secretion (pancreas-in response to glucose) glucose uptake (muscle) bone resorption fibroblast proliferation collagen synthesis changes in mood and excitability altered leukocyte functions (anti-inflammatory)
Glucocorticoid anti-inflammatory mechanisms • leukocyte traffic control • leukocyte function • inhibition of the prostaglandin/leukotriene pathway
GC REC GC GC blocks NF-kB function and may also induce inhibitors of the NF-kB pathway like IkB
Glucocorticoids block neutrophil migration out of blood vessels by inhibiting response to chemotactic molecules and by preventing passage of neutrophils through endothelial gap junctions
Glucocorticoids inhibit macrophage-Tcell interactions by blocking interleukin induction and preventing macrophage activation
Glucocorticoids inhibit tissue destruction by macrophage by blocking both macrophage activation and subsequent release of proteases like collagenase, elastase, and plasminogen activator
Toxic effects of chronic glucocorticoids gluconeogenesis (liver)--hyperglycemia release of amino acids-catabolism (muscle)--muscle weakness release of fatty acids-lipolysis (fat)--together with increase in insulin, leads to inappropriate fat deposition, obesity insulin secretion (pancreas-in response to glucose) hyperinsulinemia bone resorption--leading to osteoporosis, fractures fibroblast proliferation--thin skin, bruising, poor wound healing collagen synthesis growth retardation (in children) changes in mood and excitability--euphoria, restlessness altered leukocyte functions (anti-inflammatory)--may mask underlying symptoms suppression of the HPA axis: acute withdrawl can lead to death
Other members of the nuclear receptor family AF2 AF1 a b LXR oxysterols FXR farnesoids PXR zenobiotics CAR phenobarbital LXR and FXR: regulate cholesterol and bile acid homeostasis PXR regulates drug metabolic (cyp)genes