Download

1 / 18
200 likes | 982 Views
HEMOCROMATOSIS PORFIRIA. HOMEOSTASIS DEL HIERRO. - Absorcion duodenal en forma ferrosa a través del transportador de metal divalente (DMT1) (1 mg/día en varón y 1.5 en mujer) - Puede almacenarse en la célula como ferritina o salir de la célula

E N D
HEMOCROMATOSIS PORFIRIA
HOMEOSTASIS DEL HIERRO - Absorcion duodenal en forma ferrosa a través del transportador de metal divalente (DMT1) (1 mg/día en varón y 1.5 en mujer) - Puede almacenarse en la célula como ferritina o salir de la célula por el transportador ferroportina. En el plasma se fija a la proteina transportadora (transferrina) pero antes, la ceruloplasmina lo oxida - Los hepatocitos adquieren Fe por el receptor de transferrina - Los macrofagos del SER la adquieren por fagocitosis de eritrocitos viejos y la liberan por ferroportina - Como reacción al aumento de Fe, el higado produce hepcidina cuyo déficit produce un cuadro similar a la hemocromatoris. - El gen HFE codifica una proteina de membrana similar a MHC1 que forma un complejo con la microglobulina β2 y el receptor de transferrina, su déficit aumenta la afinidad de dicho receptor con lo cual aumenta la captación celular de Fe
INTERACCION FISIOLOGICA HEPCIDINA-FERROPORTINA CELULA Hierro Ferroportina Ferroportina Plasma Hepcidina Fe Fe • La ferroportina es una proteina de membrana que saca Fe de la celula • La hepcidina circulante(proteina hepatica se une a la ferroportina • Ambas se internalizan degradándose la ferroportina, por ello disminuye • la salida celular de Fe
HEMOCROMATOSIS Hemocromatosis: Exceso de absorción intestinal de hierro que se deposita en hígado, páncreas, corazón e hipofisis fibrosándolos y alterando su función. El pigmento depositado se llama hemosiderina Debe diferenciarse de la hemosiderosis (depósito de hierro secundario a administración de hierro o trasfusiones)
ESTADOS DE SOBRECARGA DE HIERRO Hemocromatosis hereditaria (autosomica recesiva) • Relacionada con HFE (tipo 1) • No relacionada con HFE - Hemocromatosis juvenisl (tipo 2) - Mutación del receptor 2 de transferrina (tipo 3) - Mutación del gen de ferroportina (tipo 4) - Mutación del gen de Ferritina (tipo 5) Sobrecarga adquirida de hierro (hemosiderosis) a) Hematológicas: • Anemias: Talasemia Mayor, Sideroblástica, Hemolítica • Trasfusiones (sobrecarga de hierro) b) Hepatológicas • Hepatitis C, cirrosis alcoholica • Esteatohepatitis no alcoholica • Porfiria cutánea tarda c) Otras: • Deficiencia de Hepcidina(proteina hepatica que favorece la deplección intracelular del Fe) • Aceruloplasmina hereditaria • Ingesta dietética de Fe
HEMOCROMATOSIS HEREDITARIA Mutación del gen HFE con aumento de la afinidad del receptor de transferrina, (no bien conocido porque aumenta absorcion de Fe) - Más frecuente en hombres. 1/10 personas es portador heterozigota - Hay también déficit de hepcidina Patogenia - El acúmulo de hierro eleva la sideremia, luego la transferrina (transportadora) luego la ferritina (depósito) - En fase avanzada hay más de 20 grs de hierro depositados en las células lo cual rompe los lisosomas, peroxida lipidos de las membranas y activa la síntesis de colágeno Anatomía patológica - Depósito de hierro en hígado, páncreas, corazón, glándulas endocrinas, piel (pigmentación melánica) sinovial (cristales de pirofosfatos) - Nódulos hepáticos con fibrosis perinodular y depósito de Fe en cels de conductos biliares, Kupffer. Proliferación de conductos biliares. Al final produce una cirrosis macronodular
FISIOPATOLOGIA DE LA HEMOCROMATOSIS 1 2 Y 3 Defecto cuantitativo en la interacción hepcidina –ferroportina 1.- Descenso de hepcidina circulante 2.- Menor unión a ferroportina 3.- Menor degradación intracelular de ferroportina 4.- Aumento de ferroportina en la membrana 5.- Aumento de liberación de hierro
CLINICA DE LA HEMOCROMATOSIS DESARROLLO PROGRESIVO: Grado 0-: Sin datos clínicos ni bioquímicos Grado 1.- Aumenta la saturación de la transferrina ( > 45%) con ferritina normal (< 300 mg/l en varón y < 200 en mujeres) Grado 2.- Aumento de ambas sin manifestaciones clínicas Grado 3.- Igual pero con síntomas que afectan a la calidad de vida (astenia, impotencia artropatía) Grado 4.- Con lesiones parenquimatosas que aumentan la mortalidad (cirrosis, miocardiopatía DM)
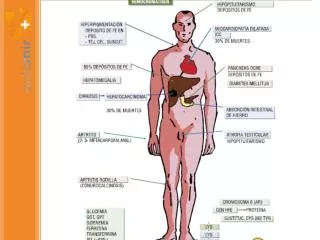
CLINICA DE LA HEMOCROMATOSIS Astenia, pérdida de peso Pigmentación melanica cara, cuello, brazos, manos piernas y genitales (90%). No suele pigmentarse las mucosas Dolor abdominal Hepatopatía (95%). Riesgo de hepatocarcinoma (30%) Diabetes (65%) por RI Artropatía (25-30%) en las manos, muñeca, codos y rodillas puede haber ataque agudo por depósitos. Puede haber quistes subcondrales Cardiopatía (15%): Cardiomegalia, arritmia, ICC Hipogonadismo hipotalámico: impotencia,atrofia testicular Otras alteraciones endocrinológicas - insuficiencia suprarrenal - hipotiroidismo - hipoparatiroidismo
HEMOCROMATOSIS Diagnóstico - Sospecha clínica - Analítica: Fe (>170 µg), saturación de transferrina (> 62%), Ferritina en plasma elevada - Biopsia hepática (medir el hierro depositado) - CT o MR hepática (aumento de la densidad) - HLA familiares (haplotipo idéntico en los homozigotos) - Test de desferroxiamina (inhibe la captación de Fe) Tratamiento - Flebotomía: 500 cc/dos veces/ semana/ dos años. luego cada tres meses - Desferroxiamina quela 10 a 20 mg/día cuando la flebotomía no está indicada
PORFIRIAS • Concepto • Patología producida por depósito de porfirinas o de • sus precursores en el hígado o en los eritrocitos • - Se produce por alteración en la síntesis del heme • - Puede ser genética o adquirida • Clasificación: • Hepáticas(ALA y PFG producidas en hígado) • - Déficit de ALA deshidratasa • - Porfiria aguda intermitente • - Porfiria cutánea tarda • - Coproporfiria hereditaria • - Porfiria variegata (déficit protooxidasa) • Eritropoyéticas • - Porfiria eritropoyética congénita (déicit urosintetasa) • - Protoporfiria eritropoyética (déficit ferroquelatasa) • - Anemia sideroblástica unida al cromosoma X
SINTESIS DEL HEM mitocondria ( - ) Glicina HEM Fe Protoporfirina IX Succinil Co. A Protoporfirinógeno IX ALA (x2) Porfobilinógeno Uroporfirinógeno IIICoproporfironógeno III Drogas, estrógenos 7 (+) 5 ALA sintasa 4 coprooxidasa citoplasma 1 ALA deshidratasa HMB sintasa 2 urodecarboxilasa 3 urosintasa 6 Los números indican la localización del defecto en cada uno de los procesos
PORFIRIA POR DEFICIT DE ALA DESHIDRATASA 1 Autosomica recesiva. Los pacientes heterozigotos son asintomáticos Clínica: - Similar a la porfiria aguda intermitente - Dolor abdominal y neuropatía Diagnostico: - Elevación de ALA urinario y coproporfirina. Tratamiento: - Como la porfiria aguda intermitente.
PORFIRIA AGUDA INTERMITENTE (PAI) 2 Déficit HMB sintasa mayor al 50%. Autosómica dominante Clínica: Heterozigotos pueden ser asintomáticos. Evoluciona en crisis con un desencadenante (alcohol, dietas estrógenos, barbituricos, sulfamidas) - Dolor abdominal con ileo, náuseas, vómitos y estreñimiento - Neuropatía periférica: Motora, sensorial (dolor) o de pares craneales (parálisis respiratoria o bulbar). Convulsiones - Afectación parasimpática: Taquicardia, parada, hipertensión - Afectación mental (ansiedad, depresión, alucinaciones, paranoia) - Hiponatremia (SIADH) Diagnóstico: > ALA y PGB en plasma y orina en el ataque Tratamiento: Narcoticos para el dolor. Heme 3-4 mgrs iv 4 días como hematina, albúmina o arginato Prevención: Evitar factores desencadenantes
PORFIRIA CUTANEA TARDA 3 Es la más frecuente Déficit de urodecarboxilasa hepática Tipos: a) Esporádica (enzima normal en eritrocitos) b) familiar (tipos 2déficit también en eritrocitos Desencadena: Alcohol,estrógenos, hexaclorobenceno Clínica: - Fotosensibilidad cutánea (vesiculas y bullas en áreas expuestas) - Hipertricosis, hiperpigmentación. - Lesión hepatica (carcinoma) Diagnostico: - > de porfirina hepática, plasma, orina y heces. - Coproporfirina elevadas en heces. Tratamiento: Flebotomías (500 cc cada 1-2 semanas). Cloroquina o hidroxicloroquina Profilaxis: Evitar tomar el sol
COPROPORFIRIA HEREDITARIA 4 Déficit de coprooxidasa. Autosómica dominante Clínica: Neurovisceral similar a la PAI y cutánea como la PCT Diagnostico: - Elevación de Protoporfirina, Coproporfirinas ALA y PBG en heces y orina durante la crisis. Tratamiento: Hematina en las crisis. Evitar el sol PORFIRIA VAREGATA 5 Déficit de protooxidasa. Autosomica dominante Clínica: Neurovisceral o fotosensibilidad. Diagnóstico: - Elevación de Protoporfirina y coproporfirina en heces y orina. Elevación de porfirinas plasma Tratamiento: - Hematina durante la crisis
ANEMIA SIDEROBLASTICA UNIDA AL CROMOSOMA X 6 Autosomica recesiva por déficit de urosintetasa Clínica: - Anemia hemolítica. (deposito en eritrocitos e hiperesplenismo) - Fotosensibilidad en piel (bullas, vesiculas, - Hipertricosis - Pigmentación focal Diagnostico: - Elevación de copro y uroporfirinas en medula, eritrocitos orina y heces Tratamiento: - Trasfusión. - Esplenectomía. - Evitar la luz.
PROTOPORFIRIA ERITROPOYETICA Déficit de ferroquelatasa. Autosómica dominante. Depósito de protoporfirinas en eritrocitos y eliminación por bilis y heces. Clínica: - Fotosensibilidad en piel: enrojecimiento y picor tras el sol - Liquenificación en piel, estrías en labios y uñas. - No hay hemólisis - Puede haber litiasis o insuficiencia hepática con muerte Diagnostico: - Elevación de Protoporfirina en medula, eritrocito, plasma. bilis y heces. Tratamiento: - Betacarotenos oral para el picor (120 a 180 mg/día) - Colestiramina. - Esplenectomía si hay hiperesplenismo. - Dar hierro si hay déficit