Download

1 / 37
470 likes | 1.83k Views
Sclerosi Laterale Amiotrofica (SLA) . Malattie del neurone motore Spinal Muscolar Atrophyes (SMAS) (Motor Neuron Diseases MND) Malattie dei motoneuroni (SLA) (Motor Neuron Disease). Spinal Muscolar Atrophyes (SMAS) Forme infantili. Forme infantili prossimali (AR)

E N D
Malattie del neurone motore Spinal Muscolar Atrophyes (SMAS) (Motor Neuron Diseases MND) • Malattie dei motoneuroni (SLA) (Motor Neuron Disease)
Spinal Muscolar Atrophyes (SMAS)Forme infantili • Forme infantili prossimali (AR) Tipo I (acuta severa, Werding Hoffmann) Tipo II (sub-acuta, Intermedia) Tipo III (lieve, Kugelberg-Welander) • Forma infantile prossimale AD • Forma infantile distale AD o AR • Forma infantile bulbare (Fazio-Londe) (AR) • Forma bulbare (S di Brown- Vialetto van Leare) (AR) • SMA con Oftalmoplegia (AR) • SMA scapolo-omerali (AR) • SMA con Ritardo Mentale (AR)
Spinal Muscolar Atrophyes (SMAS)Forme adulte • SMA cronica prossimale (AD o AR) • Neuropatia bulbo spinale X-linked • SMA distale
Malattia dell’età adulta che che interessa i neuroni motori spinali, bulbari e corticali a decorso progressivo ad esito infausto ed eziologia sconosciuta.
Varianti cliniche • Forma classica o malattia di Charcot (SLA) • paralisi bulbare e pseudo bulbare progressiva (PBP) • Forma pseudopolinevritica (di Patrikios) • Atrofia muscolare progressiva (AMP)
Epidemiologia • Prevalenza 3-7 casi per 100,000 abitanti • Incidenza 1 caso per 100.000 abitanti per anno (0.4-1.8) • Distribuzione uniforme • 5-10% casi malattia ereditaria con trasmissione AD (raramente recessivo) • Età di insorgenza: 50-60 anni • M/F = 1.6/1 • Durata media di malattia = 3 anni
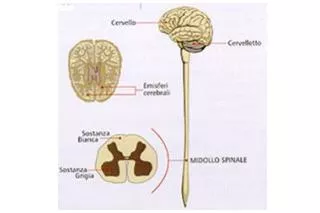
Quadro neuropatologico • Marcata perdita neuronale ed atrofia delle radici anteriori, dei rigonfianti midollari, dei nuclei di numerosi nervi cranici e delle circonvoluzioni anteriore del lobo pre-centrale • Istologicamente: Perdita assonale con alterazione della guaina mielinica e dal punto di vista ultrastrutturale, accumulo di di neurofilamenti al livello delle corna anteriori (corpi sferoidei)
Quadro neuropatologico • Atrofia delle radici anteriori • Atrofia dei rigonfianti midollari • Depauperamento dei motoneuroni delle corna anteriori del Midollo • Depauperamento dei motoneuroni periferici di numerosi nervi cranici (IX, X, XI, XII, VII, V) • Degenerazione del fascio piramidale • Atrofia del lobo frontale al livello della corteccia motoria • Nervi periferici: Atrofia dell’assone e sofferenza della sostanza bianca • Atrofia muscolare Istologicamente: • Corpi sferoidali • Corpi di Bunina • Fasci gliali
Ipotesi eziologiche • Ipotesi virale • Ipotesi tossica (piombo, mercurio) • Ipotesi Autoimmune • Ipotesi dello stress ossidativo • Ipotesi della intossicazione da neurofilamenti • Ipotesi eccitotossica
Ipotesi immunitaria • Trasferimento delle IgG sieriche dall’uomo al modello animale • Incremento della liberazione di Ach alla terminazione Neuromuscolare Il rilascio di Ach dipende dai canali del Ca Voltage-sensitive e dall’aumento della concentrazione del Ca nella terminazione presinaptica • Identificazioni di anticorpi diretto contro i canali del dal Ca voltage-sensitive nel 75% dei pazienti
SOD-1 • In alcuni casi di fSLA (20%)presenza di una mutazione del gene che codifica per la proteina enzimatica Cu-Zn superossidodismutasi sul Cromosoma 21 q22.1-22.2 • Cu-Zn superossidodismutasi è un enzima in grado di catalizzare la dismutazione dell’anione superossido in ossigeno e perossido di idrogeno. • Un difetto di tale enzima lascia via libera all’accumulo di radicali tossici in grado di determinare la morte cellulare (Gain of function)
Possibili meccanismi 1. La SOD-1 mutata può accettare substrati come il perossinitrito (ONOO) con formazione di nitronium che può donare gruppi di nitrato alle tirosine. 2. Può interagire con H2O2 portando alla formazione di radicali idrossilici. 3. Potrebbe acquisire proprietà pro-apoptotiche 4. Potrebbe disturbare la sintesi di RNA trasfer 5. Formazione di aggregati di proteine mutate e proteine della cellula con formazione di precipitati 6. Alterare l’attività della calcineurina, fosfatasi sotto il controllo del Ca e della calmodulina
Neurofilamenti • In alcuni casi di SLA sporadici: alterazione di una regione del cromsoma 11 che codifica per la subunità H dei neurofilamenti. (fattore predisponenete) • Accumuli di neurofilamenti sono un reperto patologico frequente nella SLA • Presenza di accumuli di filamenti nei motoneuroni spinali anche nel Wobber mouse, modello animale della SLA .
Relazione tra la mutazione SOD-1 e l’accumulo di neurofilamenti • Riscontro di accumulo di neurofilamenti in un caso di fSLA con mutazione SOD-1 • Inclusioni di neurofilamenti nel topolino transgeniche esprime mutazione SOD-1 • Le proteine dei neurofilamenti potrebbero essere il bersaglio della SOD-1 mutata attraverso alterazioni di fosforilazione delle proteine dei neurofilamenti da parte della calcineurina.
Ipotesi eccitotossica Eccesso di trasmissione eccitatoria glutammatergica determina un accumulo di Ca++ intraneuronale e conseguente morte cellulare • Concentrazioni di glutammato maggiori nel siero e nel liquor di pazienti affetti da SLA. • Difetto di reuptake gliale del glutammato legato alla perdita di una proteina trasportatrice GLT-1 espressa sulla superficie delle cellule astrogliali. • Assenza nei motoneuroni delle proteine calbindina e parvalpumina in grado di tamponare gli ioni calcio e di proteggere quindi i neuroni dalla tossicità mediata dal recettore AMPA-Kainato
Forma classica o malattia di Charcot CLINICA 1. Lesione del motoneurone periferico (nuclei dei nervi cranici motori somatici e corna anteriori del midollo) paresi e atrofia muscolare 2. Lesione del motoneurone centrale (corticale) paresi con accentuazione dei riflessi propriocettivi e spasticità0
Lesione del motoneurone periferico • Presenza di fascicolazioni che precedono il disturbo trofico • Paresi ed atrofia spiccata più frequentemente nei mm. delle mani (mano ad artiglio, a scimmia o scheletrica). • Disturbi trofici e paretici dei muscoli della faccia, della lingua, della masticazione, della faringe e della laringe con gravi disturbi della deglutizione, della masticazione e disartria
Lesione del motoneurone centrale • Concorre nel provocare fenomeni paretici • Determina aumento dei ROT, presenza di Babinski, cloni della rotula e del piede, etc. • Spasticità • L’evocabilità dei segni legati alla compromissione del motoneurone centrale dipende dal grado di compromissione del MN periferico
Quadro clinico comune in fase intermedia • Segni di lesione periferica agli arti superiori (atrofia, paralisi flaccida, fascicolazioni ed areflessia) • Segni di lesione centrale (piramidale) agli arti inferiori (spasticità, iperreflessia, normale trofismo muscolare) • Segni di lesione periferica nei nervi cranici, soprattutto bulbari (IX-X-XI-XII) con paralisi labio-glosso-laringea.
Elettromiografia Segni di sofferenza muscolare neurogena mielopatica 1. Aumentata attività spontanea • Potenziali di fascicolazione • Potenziali di fibrillazione 2. Contrazione volontarie massimale • Riduzione del numero di UM • Modificazione dei parametri dei singoli potenziali di UM: lunga durata, polifasici, di grande ampiezza per i processi di reinnervazione collaterale ad parte dei motoneuroni normali(sprouting)
EMG Le alterazioni devono essere ricercate in almeno: • 1 muscolo della regione bulbare: (facciali, masticatori o lingua) • 1 muscolo della regione toracica (paraspinali, parasternali, addominali) • 2 muscoli delle regioni spinali cervicale e lomabosacrale innervati da due radici diverse
Criteri diagnostici El Escorial 1998 (WFN) Evidenza di: • Interessamento del motoneurone inferiore (LMN) (clinica, elettrofisiologica, patologica) • Interessamento del motoneurone superiore (UMN) (clinica) I segni di interessamento motoneuronale deve essere cercato in almeno 4 livelli (tronco-encefalo, cervicale, toracica e lombosacrale) Assenza di • Immagini in grado di spiegare i segni clinici ed elettrofisiologici
Criteri diagnostici El Escorial 1998 • Definita: segni-sintomi di deficit motoneuronali a carico di 3 aree (tronco, midollo cervicale, midollo dorsale e/o lombosacrale) • Probabile: segni-sintomi di deficit motoneuronali a carico di 2 aree e quelli da motoneurone centrale sono al livello più rostrale rispetto a quelli da motoneurone periferico. • Possibile: segni-sintomi centrali e periferici sono a carico di 1 area oppure quelli ad motoneurone centrale sono a carico di 2 o di 3 aree • Sospetta: segni-sintomi da motoneurone periferico sono a carico di 2 o 3 aree.
Altre indagini neurofisiologiche • Velocità di conduzione sensitive e motorie, nella norma • Stimolazione ripetitiva a bassa frequenza può evidenziare un precoce decremento di ampiezza del potenziale di risposta muscolare per deficit a livello delle placche neruomuscolari. • Stimolazione magnetica
Altre indagini diagnostiche • Potenziali evocati motori: rallentamento del tempo di conduzione centrale (30% dei caso) • Aumenti della creatina-fosfochinasi (CPK) fino a 2-3 volte • Raramente reperti di laboratorio di incerto significati (gammopatie monoclonali, autoanticorpi anti-GM1 etc. • LCR normale, raramente modesto aumento delle proteine totali • RMN può evidenziare un’alterazione di segnale a livello dei fasci cortico-spinali tali da rendere visibile il decorso del fascio dalla capsula interna lungo tutto il tronco dell’encefalo. Possibile atrofia della corteccia motoria
Varianti cliniche • Forma classica o malattia di Charcot (SLA) • paralisi bulbare e pseudo bulbare progressiva (PBP) • Forma pseudopolinevritica (di Patrikios) • Atrofia muscolare progressiva (AMP)
Forma bulbare (paralisi bulbare progressiva) Età d’esordio ed evoluzione sovrapponibili alla forma classica. Dominano i segni di patologia bulbare. • Disartria e disfagia, con reflusso di liquidi prima e solidi poi • Disturbi della masticazione (mandibola cadente) • Presenza di segni piramidali (la differenzia dalle neuropatie) • Ipomobilità della lingua ce appare ipo/atrofica con fascicolazioni. • Si associano i disturbi dei neuroni di moto presenti nella forma classica.
Forma pseudopolinevriticaSindrome di Pierre-Marie e Patrikios • Esordio con disturbi motori e trofici agli arti inferiori • Loggia antero-laterale delle gambe (andatura steppante) • Presenza di segni piramidali che la differenzia dalle polineuropatie • Evoluzione sovrapponibile alla SLA classica con coinvolgimento degli arti superiori
Atrofia muscolare progressiva (AMP) • Coinvolgimento del distretto piramidale è ridotto o nullo • Esordio con disturbi di forza e trofismo distali (mano da scimmia) • Assenza dei riflessi osteo-tendinei • Evoluzione verso i distretti prossimali • Sopravvivenza fino 20-30 anni • Sindrome di Vulpian-Oppenheim: variante a decorso prossimo-distale
Sclerosi laterale primaria (SLP) • Rare forme piramidali pure • Marcia ipertonica • Accentuazione dei riflessi osteo-tendinici, • Clono della rotula e del piede • Paresi bulbare • Sindrome pseudobulbare Sindrome di Mills (paralisi ascendente progressiva unilaterale) • Interessamento del fascio piramidale
Diagnosi differenziale • Mielopatia spondilosica cervicale • Sindromi Neurologiche paraneoplastiche • Sindrome Post-poliomielitica
Terapia specifica • Riluzolo (Inibitore del release pre-sinaptico di glutammato). Determina un aumento della sopravvivenza di 3 mesi Nessuna efficacia hanno dimostrato i seguenti trattamenti • plasmaferesi e/o immunosoppressori • Interferone • Guanidina • Terapia chelante • TRH • Ceftriaxone
Terapia sintomatica • Affaticabilità di tipo miastenico (mestinon, guanidina) • Spasticità :(tizanidina, baclofene, diazepam, clorpromazina) • Crampi muscolari (dantrolene) • Scialorrea (anticolinergivi, antidepressivi) • Disturbi del sonno • Disfagia (Riabilitazione, sondino naso-gastrico, PEG) • Disartria (Logopedia, terapia comunicativa) • Insufficienza respiratoria (Ventilatori portatili a pressione positiva con maschere nasali o oronasali) • Terapia si supporto (Fisioterapia)
Terapia sintomatica • Informazione e supporto psicologico • Eventuale psicoterapia di supporto e trattamento con psicofarmaci antidepressivi • Effetto placebo di “neurotrofici” o polivitaminici
Terapia sintomatica: metodi invasivi proposti per i disturbi bulbari • Miotonia cricofaringea, • Gastrotomia percutanea (PEG), • Sondino naso-gastrico a permanenza • Tracheostomia e mino-tracheostomia • Funzionalità respiratoria: respiratori portatili per uso domiciliare
SLA secondarie • SLA post-poliomileitica • SLA esotossica • Malattia motoneuronale da raggi • SLA post-traumatica • SLA paraneoplastica • Condizioni disendocrine