Download

1 / 32
320 likes | 332 Views
Explore the raw data from the Nematode Genetic Analysis experiment comparing the Bristol N2 and CB4856 Hawaiian strains. Analyze the population size and starting allele frequency to assess the importance of allele frequency in genetic diversity and divergence.

E N D
Nematode prac data analysis and report GEN2EGE 2018
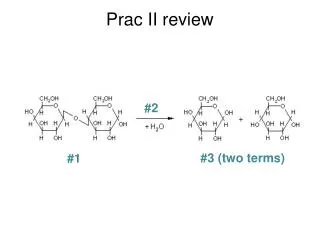
What do the raw data look like? • The difference between the two strains is in their DNA… as you have seen. • One is called Bristol N2 and the other is CB4856 Hawaiian • There are several thousand verified single nucleotide polymorphisms or SNPs between them. • Some of these change a restriction enzyme site, so that one allele retains the cleavage site and the alternative allele does not. • These are called snip-SNPs
Tuesday and Wednesday • You controlled the population size. • The second variable was the starting allele frequency. • This was set at 0.5/0.5 for Tuesday (both alleles at the same frequency), and 0.1 cut (Hawaiian) / 0.9 uncut (Bristol N2) for Wednesday. • You should analyse the two days separately, then compare in order to assess the importance of allele starting frequency.
What have we scored? • We have gone over your gels and have extracted data from those where unambiguous genotypes that could be assigned in the entire sample for a single population. We also digested more of your PCRs, re-ran some gels and added some of our own data. • The raw counts of CUT and UNCUT are on LMS, broken down by day (= starting allele frequency) and population size. • There are several populations for each combination of population size (3 & 30) and allele frequency (0.9/0.1 and 0.5/0.5). • You should use these data for your report, along with appropriate simulation output from your own AlleleA1 using the same population size and allele frequency parameters.
Hawaiii Bristol
Introduction: must include • Effective population size (Ne) • what is it? • what are the consequences, in terms of genetic drift, of reductions in Ne for • genetic diversity (loss/fixation of alleles) within populations • increase in divergence between populations • what are the other major determinants of genetic diversity? In particular, what are the effects of population bottlenecks and allele starting frequencies?
Introduction (cont’d) • Nematodes (Caenorhabditis elegans) • what are they... very brief statement about life history • why are they useful for this experiment (things like ease of culture, timing of development etc) • Clear statement of the genetic hypothesis being tested (this is NOT the null hypothesis tested in the c2 you will perform) and the Aims of the experiment (which is more than to simply test the hypothesis)
Introduction (cont’d) • the introduction is usually around 25 – 30% of the total length of a report (look at some publications.... same formula) • this means your introduction should be around 300-400 words. • It should start general (what is Neetc) and then focus more specifically on the important determinants (Ne and allele frequency), ending with a clear statement of the hypothesis you are to test and [briefly] how you intend to test it.
If in doubt, look at the paper you have been assigned to do your talk on, or the two papers that you have read recently for the paraphrasing. The introductions are about the recommended length, and they start fairly general then become increasingly focused, ending with a brief statement of the experiment to be reported (hypothesis and experimental/analytical approach).
Methods • There is nothing to do here other than “refer to the GEN2EGE practical notes. • That is all you are required to do. No more. • In particular, you are not required to note the “revisions” that were made at various stages.
Results • You should include the data table that will be on LMS. This will be the same for the whole class. • Remember each table/figure MUST have a number (Table 1, Table 2, Figure 1 etc) and a legend that says what it is. • There should be a sentence or two in the main text that summarises (in words) the data contained in the table or figure, and makes explicit reference to the table or figure. • This section should also be 25 – 30% of the total length.
AlleleA1? • As you will now know, AlleleA1 can generate a lot of output. • You must choose carefully which output to include in your report, but you must include some AlleleA1 data that illustrates the theory. • The most important criterion for choosing which output is whether that figure or table will support your discussion of the class data… “Does this AlleleA1 output explain/illustrate/complement the class data”? • You will be penalised for inappropriate inclusion of Allele A1 output.
Fixation and AlleleA1 • One feature of the simulation data that you cannot derive from the experimental data is the speed at which fixation occurs. • Fixation = one allele goes to 100% and the other is lost. • So, in your AlleleA1 data be sure to include (and describe) fixation…. How rapidly do populations go to fixation under different starting conditions of allele frequency and population size.
Data analysis in the Results • This is the bit that most students find difficult, so pay attention. • You MUST test your data statistically. • The only test we ask you to apply is the chi-square test, to determine whether (a) the genotype frequencies and (b) the number of “fixed” populations you observe are consistent with the expected values. • These tests form part of the Results section. • Note that you should record ALL of (a) the test used, (b) the test statistic C2 (c) degrees of freedom (d)and the p value estimated from the value of C2 and d.f. • It is NOT acceptable to simply report a p value without the other information.
Discussion • Your focus of your discussion will be on the statistical analysis of the class data, with reference to AlleleA1 for support where appropriate. • Some things you might consider are • What is the evidence that drift (or selection) has occurred? • Is the drift strong or weak? And is it the same in all 4 populations? • Which variable (starting allele frequency and population size) seems to be more important in determining the extent of drift? • What role, if any, might the population bottlenecks you imposed have played?
How to write a Discussion • The Discussion usually starts with a brief restatement of the hypothesis and the experimental approach. • You then identify the main influences on genetic drift and proceed to discuss your data (and the stats) in a logical order that reflects the main influences • These are, of course, population size and allele starting frequency! You may use subheadings if you think this will help, but it is not mandatory.
Always end by integrating the two parts • Lastly, you must “zoom out” again • To do this, first integrate your own Discussion into a statement of how population size and allele starting frequency interact. • Then zoom out further to say how your observations (class data + stats) fit or do not fit with what you understand to be the theory. This is where the AlleleA1 data are likely to be most useful to you.
And now, before we start on the statistics…. • DO NOT confuse: • the genetic hypothesis tested by the experiments viz. that effective population size and allele frequency determine the extent of genetic drift, • with the statistical hypotheses viz. that the difference between the observed and expected values [of allele frequency] differ due to chance.
Chi-Square analysis • Is the difference between what you OBSERVED and what you EXPECTED greater than expected by chance? • We compare the chi-square output with the p-value table to determine the level of significance X2 = Ʃ ( Observed - Expected )2 -------------------------------- Expected
X2, p-values and degrees of freedom Large x2 = difference is significant and not due to chance Small x2 = difference due to chance Degrees of freedom = (# of data columns – 1) * (# of data rows – 1) EG. A 2x2 table = (2-1)*(2-1) = 1*1 = 1 degree of freedom A 4x3 table = (4-1)*(3-1) = 3*2 = 6 degrees of freedom NOTE: if you are only analysing PART of a table, you must recalculate d.f for that part. ie. Do not use 12 d.f in a 4x5 table if you are only looking at 4 cells
Chi-Square (1) • Method 1: When you know what proportions you expect to see in your results • Works for comparing cut vs uncut ratios in the class data... you know what we started with, so use this to estimate the expected • Do this for Generation 1 and for Generation 3… or certainly at least for Generation 3 (the final generation) • (note... in the tables that follow, “cleaved” = cut)
We will use this to determine if there are MORE or LESS of a particular genotype at generations 1 & 3, compared to the starting population frequencies. • What are you testing? • NULL HYPOTHESIS: there will be no [statistically significant] change in the genotype frequency between generation 0 and generations 1 & 3 respectively
Chi-Square (1) Observed Expected Add up what you observed for each category Calculate your expected values for each category by multiplying the starting percentage, ie. 50%, by the total number
Chi-Square (1) Observed Expected Expected
Chi-Square (1) Observed Expected X2 = Ʃ ( 1103 - 1483)2 -------------------------------- 1483 X2 = Ʃ ( Observed - Expected )2 -------------------------------- Expected X2 = Ʃ ( 144,400) -------------------------------- 1483 X2 = 97.37 ..... ...... • You need to repeat this for each combination of Observed and Expected values, within • each table, and for the 4 tables • For each table, add each individual X2 values together, and check the X2 p-value table to see if • there is a significant difference • If YES, can you determine which cell or cells were the cause of the deviation? HINT: this will • help you describe and interpret your results in your report
Chi-Square (2) • Method 2: When you do not know what you expect to see in your results • Contingency table or Fisher’s Exact Test • We will use this to determine if there are MORE or LESS samples that have gone to fixation in: • Small vs large populations • 50:50 vs 90:10 • And to determine if all the populations for a given treatment (pop size & allele frequency) have done the same thing, whatever that “thing” might be i.e. have all populations behaved in the same way.
Observed Expected??? Enter data Calculate row and column totals Use row and column totals to determine the expected values using the formulas
Chi-Square (2) Observed - Example Expected
Observed Expected X2 = Ʃ ( Observed - Expected )2 -------------------------------- Expected • Like before, complete the X2 for each observed and expected combination, ie. 12 times • Add all X2 values and compare to the p-value table • If significantly different, which Obs/Exp combinations are generating the significant • change, and determine if there are MORE or LESS worms present than expected in • those categories.
Puzzled? • OK… alot to take in, I know. • So, all of this is in on LMS with explanation.