Download

1 / 18
180 likes | 198 Views
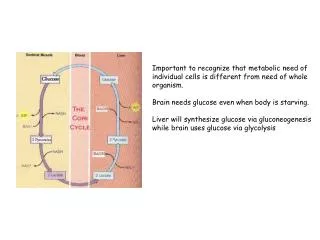
Landau free energy density. For large N we expect thermodynamic limit t=(T-T c )/T c (t=0 corresponds to the critical temperature Can think of this as averaging over many spins, ignoring spatial correlations/fluctuations …. like mean field theory. Minima of the Landau free energy density.

E N D
Landau free energy density For large N we expect thermodynamic limit t=(T-Tc)/Tc (t=0 corresponds to the critical temperature Can think of this as averaging over many spins, ignoring spatial correlations/fluctuations …. like mean field theory
Minima of the Landau free energy density For H=0, t>0, we find For H=0, t<0, we find t=(T-Tc)/Tc (t=0 corresponds to the critical temperature)
Minimization using Metropolis Monte-Carlo…. Take H=0 and kBTc=1 (L in units of kBTc) Determine using Metropolis algorithm as a function of T What is the effect of changing N? Notice that T=Tc(t+1) a=1 and b=1 in definition of L
Coding it up… declarations IMPLICIT NONE INTEGER, PARAMETER :: Prec14=SELECTED_REAL_KIND(14) INTEGER , PARAMETER :: mcmax=100000,N=1000,mcmin=mcmax/2 INTEGER :: mc,nt,ntot REAL(KIND=Prec14) :: l1,l2,eta,etalast,prob,rnd,bb,aa,t REAL(KIND=Prec14) :: etaaverage,etaanalytic REAL(KIND=Prec14), PARAMETER :: a=1,b=1,tsteps=100 REAL(KIND=Prec14), PARAMETER :: tmax=1.0d0,tmin=-1.0d0 REAL(KIND=Prec14), PARAMETER :: dtemp=(tmax-tmin)/tsteps REAL(KIND=Prec14), PARAMETER :: step=0.01d0 ! Parameters to go from Landau density to Landau free energy aa=a*N bb=b*N
Execution part… Outermost “do” loop on reduced temperature t. etaaverage and ntot are for computing average value of the order parameter do nt=1,tsteps t=tmax-dtemp*(nt-1) etaaverage=0.0d0 ntot=0 Next “do” loop is over Monte-Carlo steps (random moves).. We need to take a random step and compute Landau energy before and after step do mc=1,mcmax ! save eta, take a random step etalast=eta call RANDOM_NUMBER(rnd) l1=aa*t*eta**2+0.5d0*bb*eta**4 eta=eta+step*(0.5d0-rnd) l2=aa*t*eta**2+0.5d0*bb*eta**4
Execution part… Metropolis MC algorithm… If energy goes down, accept step… if(l2.le.l1) then ! accept the step If energy goes up, step accepted with some probability… else ! accept with some probability call RANDOM_NUMBER(rnd) prob=dexp(-(l2-l1)/(1+t)) if(rnd.le.prob) ! Accept step
Accumulate data over MC steps for averages… We need to accumulate data for each step, whether random move is accepted or not… if(mc.ge.mcmin) then etaaverage=etaaverage+eta ntot=ntot+1 endif Here, mcmin is the minimum number of MC steps before we start to accumulate data… equilibration period
Output results from MC simulation… Output results at each reduced temperature t, including average order parameter, expected value from minimum L, and the Landau free energy l2=aa*t*etaaverage**2+0.5d0*bb*etaaverage**4 if(t.ge.0) etaanalytic=0.0d0 if(t.lt.0) etaanalytic=dsqrt(-(a*t/b)) write(6,100) t,etaaverage,etaanalytic,l2 100 format(f6.3,3f12.6)
Fluctuations… N=10, fluctuations are dominant In the thermodynamic limit N--> infinity, approaches values predicted by minimum of Landau free energy For a small N, fluctuations are important: For example, for a small enough system, even for t<0 the system may periodically flip between + and - values of As N--infinity, flips between + and - not possible below t=0… ergodicity breaking…
Spatially varying order parameter: Phase-field models (r) “Coarse-grained” order parameter, which can be thought of as Gradient type term to treat domain-wall energy…and the Landau free energy functional is
Phase-field models, Monte Carlo approach This shows us how to do Metropolis MC for the case of a spatially varying order parameter Random steps in (discretized) order parameter (r) Accept/reject steps depending on Landau energy functional L[(r)] Very general, can add along with continuity equation concentrations, describe mixing of elements, diffusion, and concomitant structural phase transitions In materials science the equations are called “Cahn-Hilliard” equations, can be studied computationally
Declarations IMPLICIT NONE INTEGER, PARAMETER :: Prec14=SELECTED_REAL_KIND(14) INTEGER , PARAMETER :: mcmax=1000000,mcmin=mcmax/2 INTEGER, PARAMETER :: nn=30 INTEGER, PARAMETER :: nx=nn,ny=nn INTEGER :: ix,iy INTEGER :: mc,kt,ktot REAL(KIND=Prec14) :: l1,en,en0,K,mag,lav,mav,rnd,prob REAL(KIND=Prec14), DIMENSION(nx,ny) :: S REAL(KIND=Prec14), PARAMETER :: a=1,b=1,ksteps= 8 REAL(KIND=Prec14), PARAMETER :: kmax=3.0d0,kmin=0.5d0 REAL(KIND=Prec14), PARAMETER :: dk=(kmax-kmin)/ksteps
Initial energy l1=0.0d0 do ix=1,nx do iy=1,ny call cluster(ix,iy,nx,ny,S,K,en) l1=l1+0.5d0*en enddo enddo
MC loop, on coupling and random steps do kt=1,ksteps+1 K=kmax-dk*(kt-1) ktot=0 mav=0.0d0 lav=0.0d0 do mc=1,mcmax ! generate a random spin. Draw a number for the ix,iy of the spin to flip call RANDOM_NUMBER(rnd) rnd=rnd*dble(nx) ix=1+int(rnd) call RANDOM_NUMBER(rnd) rnd=rnd*dble(ny) iy=1+int(rnd) call cluster(ix,iy,nx,ny,S,K,en0) ! flip the randomly chosen spin and compute new energy S(ix,iy)=-S(ix,iy) mag=mag+2.0d0*S(ix,iy)/(nx*ny) call cluster(ix,iy,nx,ny,S,K,en)
Cluster subroutine subroutine cluster(ix,iy,nx,ny,S,K,en) INTEGER, PARAMETER :: Prec14=SELECTED_REAL_KIND(14) INTEGER :: ix,iy,nx,ny,l,m,n REAL(KIND=Prec14), DIMENSION(nx,ny) :: S REAL(KIND=Prec14) :: K,en ! compute interaction of spin ix,iy with its neighbors en=0.0d0 do n=0,1 l=ix-1+n*2 m=iy ! implement the pbc if(l.lt.1) l=nx if(l.gt.nx) l=1 en=en-K*S(ix,iy)*S(l,m) enddo do n=0,1 l=ix m=iy-1+n*2 ! implement the pbc if(m.lt.1) m=ny if(m.gt.ny) m=1 en=en-K*S(ix,iy)*S(l,m) enddo return end