Download
1 / 28
280 likes | 798 Views
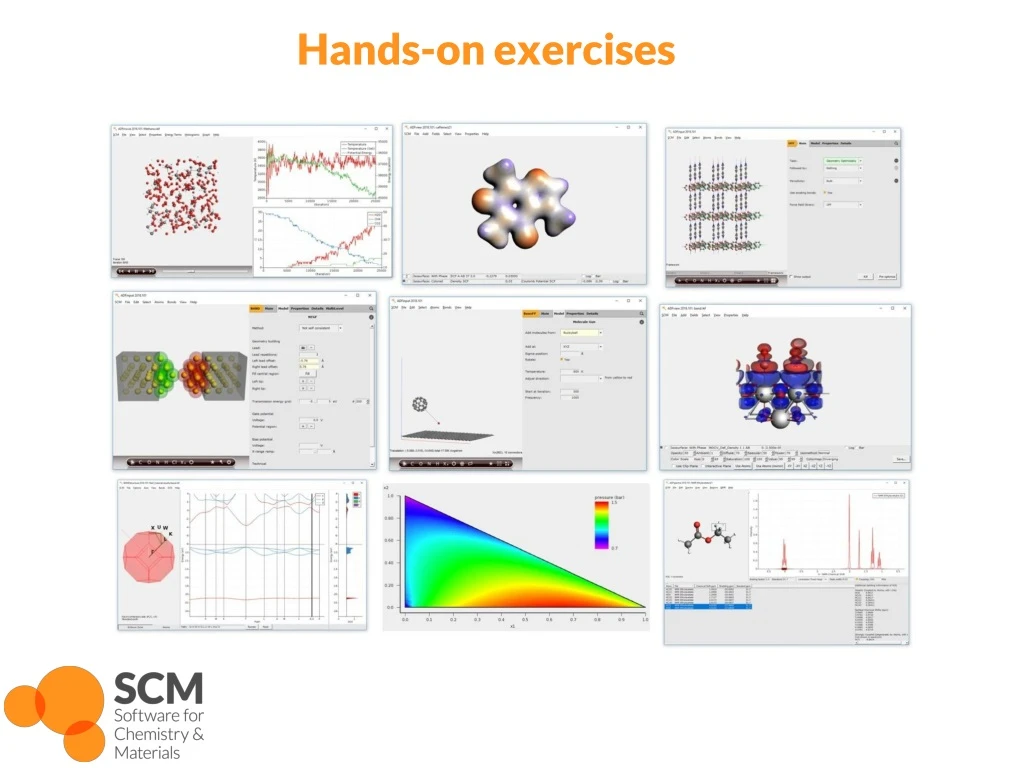
Hands-on exercises. Getting started with the GUI. Starting ADFjobs : job bookkeeping tool Win: dbl -click desktop item Mac: open Application Linux: run $ADFBIN/ adfjobs Other GUI modules: (Input, View, Levels, Movie, Spectra, Band Structure, ADFTrain , COSMO-RS, … )

E N D
Getting started with the GUI Starting ADFjobs: job bookkeeping tool • Win: dbl-click desktop item • Mac: open Application • Linux: run $ADFBIN/adfjobs • Other GUI modules: (Input, View, Levels, Movie, Spectra, Band Structure, ADFTrain, COSMO-RS, …) • Can be opened by dbl-clicking ‘.exe’ (Win) or opening e.g. ‘$ADFBIN/adfinput’
ADFjobs: job bookkeeping switch GUI functionality define & switch queues reports & templates change default e.g. cores / nodes job status see files for this job queue search all jobs / folder view
Basic calculations & settings switch modules search job types & set up job type charge/spin functional & relativistic appr basis & numerical accuracy builder tools preoptimize > = more details symmetryze
Building molecules www.scm.com/doc/Tutorials/GUI_overview/Building_Molecules.html • NB: tutorials also offline! • Import: SMILES, xyz, cif, pdb, … • Included library + building • Excercise: Build acetophenone • By searching for it in the GUI • By starting from the benzene template (press 2 for double bond, Ctrl+E to add Hs) • By importing smiles CC(=O)c1ccccc1 (e.g. from Wikipedia or Chemspider) • Exercise: Symmetrize, pre-opt (MOPAC, DFTB) • Optimize with ADF: SR-ZORA-PBE-D3(BJ)/DZP – differences? speed?
Quick properties with COSMO-RS • From the SCM menu, choose COSMO-RS • Add the acetophenone smiles string • Properties => Pure compound properties • Other properties (vapor pressure, solubility (install database), logP, … ) • Results should be better with MOPAC or ADF calculations of compounds
Spectra: IR www.scm.com/doc/Tutorials/ADF/ADF-GUI_tutorials.html#spectroscopy • Excercise: Calculate & visualize frequencies • First optimize geometry, or compound job ADF/AMS • Try ADF, DFTB3-D3BJ/3-ob, GFN-xTB, MOPAC • NB analytical frequencies for most GGAs, not for hybrids • Go to spectra, visualize the CO stretch at ~1690cm-1 • Increase the line width to ~20 & compare to NIST data • Add spectra of other calculations (File -> Add) 1.3 2. 1 2 or
Spectra: UV/VIS • Exercise: • With ADF: calculate 10 allowed excitations • use SAOP model potential, DZP (or TZP), no core • See also UV/VIS FAQ for more tips • Go to spectra, change x-axis to nm • Increase the line width to ~10 • Visualize the pi-pi* NTOs at ~250 & 285nm • Compare to NIST data • Now rerun with method ‘sTDA’ and tick TDA • Also try TD-DFT+TB (ADF) • and TDDFTB (DFTB3-D3BJ/3-ob, QN2013, GFN-xTB) • Compare timings & spectra (File -> add spectra)
Band structure, pDOS, fat bands, COOP • Exercise: ZnS bulk • New input, go to BAND • click on the ‘crystal’ builder tool in the bottom • select cubic -> Zincblende and accept the default • Settings: BP, SR-ZORA, and DZP • Select DOS and Bandstructure (default interpolation) • Run it!
Band structure, pDOS, fat bands, COOP • Exercise: ZnS bulk • Visualize the band structure (SCM Menu). You will automatically see the pDOS and ‘fat bands’ • ZnS is a direct band gap semiconductor (p-s transition) • Check the logfile and output for band gap info and kmesh • Low band gap: try model potentials (TB-mBJ, GLLB-sc, GGA-1/2, HSE06? (benchmark study) • Should also be converged wrtkpoints, basis, etc. • Restart the calculation from SCF and in the DOS details tick ‘COOP’ • Visualize the crystal orbital overlap population between the Zn s and S p orbitals
Band structure, pDOS with QE • Exercise: ZnS bulk with QE • Switch from BAND to Quantum ESPRESSO (may prompt download request) • Choose the same k-mesh (5x5x5), functional and Vanderbilt pseudopotentials • You will see a similar band structure, but they aren’t colored according to character • DOS can be projected by QE
Surfaces, dielectric function • Exercise: ZnS monolayer: 2D-TDCDFT • Cut the 111 surface with the slicer tool, and choose 1 layer • From properties -> dielectric function choose NewResponse • Calculate 30 frequencies between 2-5 eV • Set the SCF convergence criterion to 0.01 and switch off the z-component • Run it (you will prompted Nosymm is used)
1D PES scan on 2D system: find TS • Exercise: chemisorption of H2 on graphene • Bond the H atoms to adjacent C atoms • (Partially) Pre-optimize. NB: you can select atoms to pre-optimize interactively • PES scan, increasing both C-H distances simultaneously to 1.8 A, in 8 steps, low convergence • Try find a TS, followed by frequencies. How many imaginary modes do you have? • 2? => get rid of the 2nd one. Scan 2D? Manually break the symmetry?
The molecule gun: H2 on graphene • Exercise: hitting graphene with H2 using DFTB • Use DFTB3-D3(BJ)/3ob-3-1 (you may have made a preset by now); Choose Molecular Dynamics • Make a 4x4 supercell of 1L graphene. Add H2 some 6A above surface • MD details: 2000 steps, sample every 10, T = 100K, Berendsen thermostat, 100 fs, T=100K • Keeping H2 selected, in Model -> Molecule gun, choose Add molecule; System -> New Region • Frequency 200, start at step 1 until 2000, coords sigma 3 3 0.2, rotate, energy 0.05 eV • Run & visualize move (View-> Loop)
ReaxFF: introduction • Simulate complex systems at realistic scales • Atomistic potentials: bond orders + charge update A.C.T. van Duin et al ,J. Phys. Chem. A 2001 , 105, 9396-9409. See ReaxFF intro slides
General ReaxFF rules • No discontinuities in energy or forces • No pre-defined reaction sites or types • Only 1 atom type per element
ReaxFF parameters, transferability • many elements studied • each pair needs bonded terms • validate force field • GUI checks • training data crucial • application specific • New parameters • ADF 2013: 17 sets, 19 elements • ADF 2014: 38 sets, 29 elements • ADF 2016: 58 sets, 38 elements • ADF 2017: 79 sets, 38 elements • AMS2018: 81 sets, 40 elements + Ho/El • van Duin, Goddard, others • RxFF consulting • MCFF & CMA-ES parameterization
ReaxFF: some tips • First equilibrate your system before your production run • Usually thermostats equilibrate within some dozen ps, barostats take longer • For your equilibration, save less frames (Output frequency in Details-> MD) • If equilibrated, restart (Details->Restart) or just copy-paste last geometry • Check if your system properly equilibrates => damping constants • For Berendsenbarostat, use a high damping (e.g. 2500fs) , thermostat ~100 fs is OK • For NHC thermo/barostat check oscillation and adapt tau (see also manual) • After importing a structure (cif, database, ..): relax the system • Geometry optimization with loose criteria • OR run a few psNpT trajectory with a 0.05fs time step at 5K and 0 pressure • Avoid having lattice vectors < 10 Å • See our FAQs, e.g on ReaxFF force field availability / suitability • Contact support@scm.com
The Time Scale Problem • Even with fast (reactive) FF methods, there are still time limitations! Usually/practically: increase T But: do dynamics change?
Collective-Variable driven HyperDynamics • Hyperdynamics on a self-learning bias as function of Collective Variable: • Get real dynamics without having to construct a bias potential a priori Reset bias after transition Bal & Neyts, J. Chem. Theory Comput. 11, 4545 (2015)
CVHD for pyrolysis & combustion CVHD tutorial Bal & Neyts, Chem. Sci. 7, 5280 (2016).
Optimizing ReaxFF parameters • Parameters are • interdependent • non-linear • many • not always physically interpretable • Highly complex global optimization problem
ReaxFF: reparameterization Refine ReaxFF parameters for cross-linking polymers • Build your training set (trainset.in & geo) • Add geometries • Add conformers, trajectories • Add bond scans Most can now be done in GUI • Run CMA-ES optimization • Test errors, cross-validate • Try to further refine • See also: Co training set • Relative crystal energies • Equation of State, elastic tensor • Cohesive energy • Defect, adsorption energy • Surface energies
Some notes on (Windows) scripting Use help -> command-line and type sh to go to a Windows shell with ADF environment variables set. We now have a basic shell in which can do so some scripting Functions we will use a lot: cat, ls, pwd, various commands inside $ADFBIN
Some scripting examples cat dog output contents of the file named dog to the screen cat file > file2 output contents of file to a new file, file2 cat file2 >> file3 output file2 and append to file3 cat *.bgf > geo output all files ending in .bgf to a new geo file cat geo >> ../geo output geo to the file geo in the directory below pwd show in which directory we are cd dog.results go one directory up to dog.results cd .. go one directory down cd - go to the directory you were in before ls show which files are in this directory ls -ltra show files in directory with more details, order to time
Some other useful shell tips arrow up / down scroll through previous commands the directory below for; do; done Loop. Example: for i in 1 2 3; do cat $i/geo >> geo; done (append 1/geo 2/geo and 3/geo to geo) !$ reuse last argument. Example: ls geo cp !$ geo.1 (== cp geo geo.1) <TAB> Autocomplete. Example: you want to copy trainset.in cp tr<TAB> will search for all files here starting with ‘tr’ if only 1: it completes to that, otherwise prints a list