Download
1 / 82
820 likes | 1.35k Views
Consultations de génétique. Interrogatoire Généalogie Examen Clinique Photographies avec le consentement des patients Examens Complémentaires. Consanguinité. 36. A. B. C. F. D. G. E. 37. AD. AR. RLX. Mito. DLX. Mito. LY. Transmission héréditaire ou génétique ?.

E N D
Consultations de génétique • Interrogatoire • Généalogie • Examen Clinique • Photographies avec le consentement des patients • Examens Complémentaires
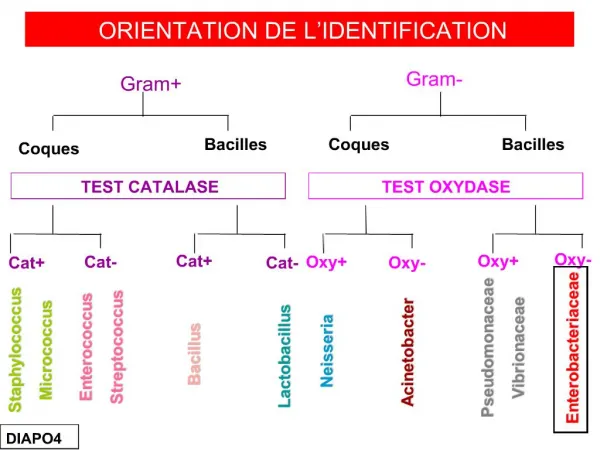
36 A B C F D G E
37 AD AR RLX Mito DLX Mito LY
Transmission héréditaire ou génétique ? L’origine génétique d’un caractère est facile à croire mais…difficile à démontrer!! Même génome mais différentes saisons ! Même génome mais différents sols ! (bleu dans un sol avec pH acide)
Famille 1: Quelle ségrégation ? Hypothèse: lié au chromosome X Une transmission père fils est incompatible avec une liaison au chromosome X
m/m m/+ m/+ m/+ m/m m/m m/m m/m m/m m/m Quelle ségrégation ? Hypothèse: Autosomique récessif Fréquence de la mutation trop importante
Quelle ségrégation ? m/+ m/+ m/+ m/+ m/+ m/+ m/+ Hypothèse: Autosomique dominant Solution la plus parcimonieuse
Quel génotype ? m/+ m/+ m/+ m/+ ? m/+ m/+ m/+ Pénétrance complète de la maladie (p=1) : +/+ Pénétrance incomplète de la maladie +/+ ou m/+ (p) (1-p)
Pénétrance La pénétrance est complète: p=1 Dans tous les cas l’individu porteur du génotype m/+ (dominante) ou m/m (récessive) est malade. La pénétrance est incomplète: p=0,8 Dans 80% des cas l’individu porteur du génotype m/+ (dominante) ou m/m (récessive) est malade. Dans 20% des cas l’individu porteur du génotype m/+ (dominante) ou m/m (récessive) n’est pas malade.
Quel risque? m/+ m/+ m/+ m/+ ? m/+ m/+ m/+
Quel risque? m/+ m/+ m/+ m/+ 1/2 [m] 1/2 [+] m/+ m/+ m/+
m/+ m/+ m/+ m/+ m/+ m/+ m/+ Famille 2: Quelleségrégation ? m/m m/m
Consanguinité Au moins un ancêtre commun La consanguinité a pour conséquence d'augmenter la fréquence des homozygotes. L'homozygotie va favoriser l'apparition de maladies récessives, si le ou les ancêtres communs étaient porteurs d'une mutation délétère récessive. Même mutation sur les deux chromosomes (paternel et maternel).
Xm/Y Xm/+ Xm/Y Xm/+ Xm/+ Famille 3: Quelle ségrégation ? Dominant lié au chromosome X - Pas de transmission père fils - Tous les filles d’un père atteint sont atteintes
Famille 4: Quelle ségrégation ? Récessif lié au chromosome X
Syndromes microdélétionnels • traits communs • Identification clinique • Leur taille est mineure (environ «3 mégabases) • Ils surviennent en général « de novo » • Le phénotype est attribué • à la perte de gènes contigus
Di George 22q11 Sd vélo-cardio-facial Insuffisance vélo-pharyngée +ou- Fente labio-palatine Dysmorphie faciale Anomalies cardiaques Hypoplasie thymique et hypocalcémie Grandes variabilités phénotypiques
Exemples Sd de Prader-Willi et Angelman -> 15q11-q12 Sd de Smith-Magenis -> 17p11.2 Sd de Williams Beuren -> 7q11.23 Sd de Miller Dieker-> 17p13.3 Sd de Di George-> 22q11 Sd Rubinstein Taybi-> 16p13.3
Sonde locus spécifique de la région 3p25 englobant le gène responsable de la maladie de Von Hippel-Lindau. Sonde marquée à la biotine détectée par immunofluoresence par la FITC, contre colorant : IP.
Cas clinique • Claire • 4 ans 1/2 • Retard des acquisitions • Marche acquise à 15 mois • Retard de langage • Troubles de l’articulation (rhinolalie) • Niveau 3 ans pour 4 ans
Microdélétion du 22 • Fréquence importante • 1/4000 • Présentation néonatale malformative • Syndrome de Di-Georges • Présentation infantile • Retard pyschomoteur modéré (60-80) • Dysmorphie subtile • Caryotype normal
Microdélétion du Chromosome 22 • Hérédité • Sporadique • Rare dominance (10%) • Locus génétique • Chromosome 22 • Microdélétion • Syndrome des gènes contigus
Dysmorphie faciale • Difficile • Petites fentes palpébrales (blépharophimosis) • Paupière entourant trop l’iris • Pas d’anomalie d’obliquité • Hyperplasie des os propres du nez • Aspect en pont du nez des nouveau-nés • Petite bouche dans le prolongement du nez • Microrétrognathie
Dysmorphie du syndrome de Di-georges • Hyperplasie des OPN • Petite bouche • Blépharophimosis • Hypertélorisme • Oreilles basses et décollées • Microrétrognathie
Présentation infantile • Retard psychomoteur modéré • Portant essentiellement sur le langage • Rhinolalie ouverte dans 50% (voile court) • Profil neuropsychologique • Enfant inhibé, timide • Langage peu utilisé (rhinolalie sévère) • Troubles psychologiques fréquent • 10% de schizophrénie
Microdélétion du Chromosome 22 • Hérédité • Sporadique • Rare dominance (10%) • Locus génétique • Chromosome 22 • Microdélétion • Syndrome des gènes contigus …..
MicrodélétionEX: Syndrome de Williams • Mathieu • 1 an • Retard discret des acquisitions • Cardiopathie • Sténose supravalvulaire de l ’aorte • Sténose Artère pulmonaire • Hypercalcémie néonatale • Dysmorphie spécifique
Génétique du syndrome de Williams • Hérédité • Sporadique • Rare dominance • Locus génétique • Chromosome 7 • Microdélétion 7q11 • Hémizygotie au locus Elastine • Délétions • Autres gènes de la région: LIM Kinase 1 …..
Syndromes acquis Syndrome de l’alcoolisme fœtal
Autres « maladies héritées », non génétiques • Syndrome d’Alcoolisme fœtal • Sous-diagnostiqué • 10 à 20% des QI 50-80 ? • 1/1000 naissances en France • Tableau malformatif • RCIU précoce • Malformations possibles • CIV, CIA • Fentes labiales ou labiopalatines
Présentation clinique • RCIU avec PC non conservé • Dysmorphie faciale • Philtrum long, lisse et bombé • Fentes palpébrales étroites … • Malformations • Hypoplasie des phalanges distales • CIA, CIV, fentes faciales
Hérédité multifactorielle • Maladies ou traits pour lesquels il existe une tendance familiale • Absence de distribution mendélienne • Combinaison de multiples facteurs génétiques et environnementaux