Download

1 / 20
200 likes | 296 Views
DNA variation in Ecology and Evolution IV- Clustering methods and Phylogenetic reconstruction. Maria Eugenia D’Amato. BCB 705:Biodiversity. Organization of the presentation. Distance ML MP. Phylogenetic reconstruction Networks Multivariate analysis.

E N D
DNA variation in Ecology and EvolutionIV- Clustering methods and Phylogenetic reconstruction Maria Eugenia D’Amato BCB 705:Biodiversity
Organization of the presentation Distance ML MP • Phylogenetic reconstruction • Networks • Multivariate analysis
Characters:independent homologous • Continuous • Discrete Binary Multistate
DNA sequence characters Alignment = hypothesizing of a homology relationship for each site Sequence comparison BLAST search - GenBank Coding sequenceblastn blastx Non-coding DNA blastn
Blast search results Score E Sequences producing significant alignments:(Bits) Value gi|87299397|dbj|AB239568.1| Mantella baroni mitochondrial ND5...101 3e-18 gi|343991|dbj|D10368.1|FRGMTURF2 Rana catesbeiana mitochondri...97.6 5e-17 gi|14209845|gb|AF314017.1|AF314017 Rana sylvatica NADH dehydr... 93.7 8e-16 The lower the E-value, the better the alignment GeneBank Accession numbers for the sequence Species that match the query
Blast search results >gi|87299397|dbj|AB239568.1| Mantella baroni mitochondrial ND5, ND1, ND2 genes for NADH dehydrogenase subunit 5, NADH dehydrogenase subunit 1, NADH dehydrogenase subunit 2, complete cds Length=10814 Score = 101 bits (51), Expect = 3e-18 Identities = 99/115 (86%), Gaps = 0/115 (0%) Strand=Plus/Minus Query 451 TTAGTTGAGGATTAAATTTTAGGATAATAACTATTCAGCCGAGGTGGCTGATGGAAGAAA 510 ||||||||||||||||||||| ||||||| ||||||||| ||||| | |||||||| | Sbjct 10203 TTAGTTGAGGATTAAATTTTAAAATAATAAGTATTCAGCCCAGGTGACCAATGGAAGAGA 10144 Query 511 AAGCTAAAATTTTACGTAGTTGTGTTTGGCTAATGCCGCCTCATCCGCCTACAAG 565 | |||| ||||||||||||||| |||||| |||| || ||||| || |||||||| Sbjct 10143 AGGCTATAATTTTACGTAGTTGAGTTTGGTTAATACCCCCTCAACCTCCTACAAG 10089 Description of the genes contained in the sequence with this Accession number Strands aligned 5’end alignment
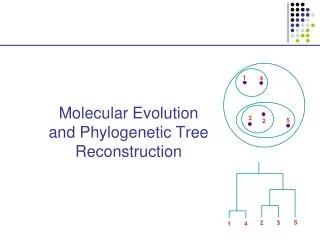
Phylogenetic reconstructionDistance methods C1 C2 C3 C4 C5 C6 C7 1 2 3 4 5 5 X 7 Distance criterion 5 x 5 Similarity / dissimilarity criterion dendrogram
Distances criterion for binary data a a + b + c a = bands common to a and b b = bands exclusive to a c = bands exclusive to b J = Jaccard’s distance P1 (x2, y2) Manhattan distance M = Euclidean distance (x1-x2) 2 + (x2-y2) 2 P2 (x1, y1)
Distance criterion for DNA data-Models of DNA susbstitution fAA fAC fAG fAT fCA fCC fCG fCT fGA fGC fGG fGT fTA fTC fTG fTT Fxy = a b c d e f g h i j k l m n o p Fxy = p = n of different nucleotides/ total n nucleotides
Models of DNA susbstitution 1 1-2P-Q 1 ln 1 4 1-2Q 1 ln 2 + dxy = D = 1 – ( a + f + k + p) Equal rate Jukes and Cantor dxy = - ¾ ln (1- 4/3 D) B = 1 – ( 2A + 2C + 2G + 2T) F81 Unequal base freqs dxy = - B ln (1- D/B) P = c + h + i + nTransitions Q = b + d + e + g + j + l + m + oTransversions K2P
Distances criterion for diploid data I Nei 1972 Jx = xi2 Jx = yi2 Jxy = xiyi Dn -ln Jxiyi JxiJyi = Cavalli Sforza 1967 Darc = (1/L) (2/)2 = cos-1xiyi
Phylogenetic reconstruction criterion for distance data Ultrametric tree (UPGMA) Additive tree (NJ) A C A V1 V1 V4 B V3 V3 V2 V2 V5 D V4 C B Properties Properties dAB = v1 + v2 dAC = v1 + v3 + v4 dAD = v1 + v3 + v5 dBC = v2 + v3 + v5 dCD = v4 + v5 dAB = v1 + v2 + v3 dAC = v1 + v2+ v4 dBC = v3 + v4 v3 = v4 v1 = v2 + v3 = v2 = v4
Maximum Likelihood 3 1 2 4 1 2 3 4 C C C A A A G G G C C C 5 + Prob……. A + Prob Lj = Prob C A A 6 LD = Pr (DH) Tree after rooting at an internal node Unrooted tree 1 J n • C….GGACACGTTTA….C • C….AGACACCTCTA….C • C….GGATAAGTTAA….C • C….GGATAGCCTAG….C L = L1 x L2 x L3…x LN. = Lj LnL = ln L1+ ln L2 + …. LN = ln Lj
Hypothesis testingLikelihood ratio test Rate variation = log L1 – log L0 Appropriate substitution Model 22 distribution d.f. = N sequences in the tree –2; or d.f = difference number of parameters H1 and H0
BootstrappingHow well supported are the groups? Trumpet fish
Maximum Parsimony Minimize tree length To obtain rooted trees (and character polarity) use an outgroup . The ingroup is monophyletic. Tree (first site) 5 changes 1 change G A • ATATT • ATCGT • GCAGT • GCCGT A G 3 1 A G G A G A 2 A 4 G
Maximum Parsimony-example Site 2 Site 3 T C A A A A C T A A C C T C C C C C Site 5 No changes Site 4 Tree length T G T T L = ki=1li T T G G T G T G
Maximum parsimony:example Sites 1 2 3 4 5 Total Tree ((1,2),(3,4)) 1 1 2 1 0 5 ((1,3),(2,4)) 2 2 1 1 0 6 ((1,4),(2,3)) 2 2 2 1 0 7 Phylogenetically informative sites
Networks • Phylogenetic representation allowing reticulation • More appropriate for intraespecific data • Ancestor is alive • hybridization, recombination, horizontal transfer, polyploidization agct 1 acat agct ac ct 2 3 4 5 7 6 acat acct agct
Multivariate clustering C1 C2 C3 C4 C5 C6 C7 1 2 3 4 5 5 X 7 • Y 2nd axis similarity criterion correlations • Z 3rd axis • • 7 x 7 • X 1st axis Calculate eigenvectors with highest eigenvalues Project data onto new axes (eigenvectors)