Download

1 / 29
290 likes | 438 Views
Analogi genów odpornościowych Oryza sativa – ich występowanie i struktura. „Everything should be made as simple, as possible but not simpler." - Albert Einstein. Grzegorz Koczyk (2004) http://www.cropnet.pl. Od sekwencji do funkcji - bioinformatyka. {A,C,G,T} n.

E N D
Analogi genów odpornościowych Oryza sativa – ich występowanie i struktura. „Everything should be made as simple, as possible but not simpler." - Albert Einstein Grzegorz Koczyk (2004) http://www.cropnet.pl
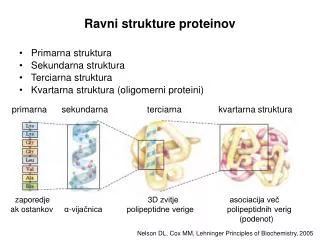
Od sekwencji do funkcji - bioinformatyka {A,C,G,T}n Funkcja (np. powielanie informacji genetycznej) {A,R,N,D,C,Q,E,G,H,I,L,K,M,F,P,S,T,W,Y,V}n/3
Homologia Podobieństwo Pokrewieństwo Homologia (podobieństwo = pokrewieństwo) Homoplazja (podobieństwo pokrewieństwo)
Porównywanie sekwencji – czy... ? NIE można bezpośrednio ocenić homologii. Możemy tylko oceniać ją na podstawie podobieństwa.
P(S>s) Porównywanie sekwencji – czy... ? • Statystyczna istotność („nieprzypadkowość”) Biologiczna istotność (homologia) E-value = P(S>s) * n Liczba oczekiwanych false positives przy przeszukiwaniu bazy liczącej n sekwencji
Indukowana odpowiedź obronna przeciwko patogenom (reakcja nadwrażliwości) Specyficzna odporność „gen-na-gen” avr R avr R Odbiór – geny R muszą umożliwiać specyficzne wykrycie konkretnego sygnału - obecności patogenu. Produkt translacji mRNA genu R odbiera sygnał – obecność produktu genu avr (czynnik awirulencji). Przekaz - geny R muszą umożliwiać wywołanie konkretnej reakcji komórki – przekaz sygnały n.p. przez aktywację kaskady kinaz.
Geny odpornościowe - struktura: Cf9 Xa21 LRR LRR SA-CC Błona komórkowa SA kinaza CC TIR-NBS-LRR CC-NBS-LRR LRR LRR Pto kinaza NBS NBS TIR CC
Geny odpornościowe – podstawowe domeny: Nucleotide Binding Site LRR Leucine Rich Repeats NBS Coiled coil / leucine zipper kinase Ser/Thr kinase CC TIR Toll / Interleukin Receptor
Ewolucja genów odpornościowych: Ewolucja– geny R muszą być zdolne do efektywnej odpowiedzi na zmieniające się czynniki w postaci produktów genów avr. Ich ewolucja powinna więc być szybka, a efektywne warianty upowszechniać się w genomie. ALE: Studia porównawcze pokazują że ortologi genów R są bardziej zbliżone do siebie nawzajem niż paralogi. Pewne geny R (Pto, RPS2) to zakonserwowane sekwencje starożytnego (w skali wieku genomu) pchodzenia.
Ewolucja genów R - równowaga: Zamiast skupiania się na efektywności pojedyńczych genów Polimorfizm na poziomie populacji Gwałtowne zmiany specyficzności genów R nadal mogą zachodzi na drodze rekombinacji międzyallelicznej. Odmienne warianty genów R obecne w puli to rezerwuar potencjalnej odporności. Indywidualne geny R są nadal zdolne do szybkiej ewolucji i gwałtownych zmian specyficzności.
Poszukiwanie genów odpornościowych in silico (uwagi): • Opisy sekwencji w bazach danych są niedokładne. Lepsze kryteria: • wysokie podobieństwo do znanych genów R (E-value < 1e-10; unikanie propagowania fałszywych diagnoz) • obecność i względne położenie domen charakterystycznych dla genów R(na podstawie przewidywanej sekwencji białkowej) Geny znalezione przez nasze poszukiwania należą do tej samej klasy strukturalnej, niekoniecznie funkcjonalnej co prawdziwe geny odpornościowe. Stąd: RGA (resistance gene analogs)
Poszukiwanie genów odpornościowych in silico (model) Baza danych sekwencji ryżu (TIGR) BLASTP na zdefiniowanym zbiorze sekwencji referencyjnych HMM („odciski palców”) domen znajdowanych w genach odpornościowych Odpytywanie powstałej struktury danych o RGA (analogi genów odpornościowych) poszczególnych klas strukturalnych. Poszukiwanie nowych wariantów.
Narzędzia – BLASTP - wyszukiwanie sekwencji: • Możliwości: • poszukiwanie podobieństwa pomiędzy sekwencjami w dużych bazach. • ocena prawdopodobieństwa przypadkowego wystąpienia dopasowania do zapytania tak dobrego jak znalezione (E-value). • Pułapki: • wyniki przeszukiwań są tak dobre jak zbiór sekwencji referencyjnych (niewykryte niewielkie homologie, problemy z wszędobylskimi sekwencjami np. kinazami). • ocena prawdopodobieństwa bazuje na pewnym modelu tworzenia losowych sekwencji. Ten model nie musi być zawsze prawdziwy. Omijamy tę wadę używając restrykcyjnych E-value (minimalizacja false positives).
Narzędzia - HMM - „odciski palców” domen: • Możliwości: • poszukiwanie domen w sekwencjach białkowych. Takie „odciski palców” są dostępne w bazach danych np. Pfam. • ocena prawdopodobieństwa przypadkowego wystąpienia dopasowania do „odcisku” tak dobrego jak znalezione. • Pułapki: • HMMy są tworzone na bazie zbioru sekwencji zawierających domenę – wyniki przeszukiwań są tak dobre jak początkowy zbiór treningowy (przykład: roślinna domena TIR). • ocena prawdopodobieństwa bazuje na pewnym modelu tworzenia losowych sekwencji. Ten model nie musi być prawdziwy. Omijamy tę wadę używając empirycznie wyznaczonych gathering thresholds.
Rezultaty przeszukiwania (BLASTP i HMMer): * Wliczając białka o fragmentarycznych NBS. ** W nawiasie liczba przypadków w których przewidziano domenę TM w prawidłowej orientacji.
Podsumowanie i uwagi: 1. Z 64574 przewidywanych przez IRGSP genów - około 2,7% (1744 sekwencje) są analogami genów R. 2. Przewidywane 597 sekwencji kodujących białka z domeną NBS, jest bardzo bliskie 600 sekwencjom uzyskanym w pierwotnym szacunku dla „draft sequence” ryżu (IRGSP, 2001). Rozkład tych sekwencji na chromosomach pokrywa się z częstościa występowania znanych genów R u ryżu. 3. Interesujące jest skupienie dużej liczby RGA zawierających domenę NBS na chromosomie 11 (28% znanych genów odpornościowych ryżu jest na chromosomie 11). „An assessment of the resistance gene analogues of Oryza sativa ssp. japonica – their presence and structure” Koczyk G., Chełkowski J. Cell Mol Biol Lett. 2003; 8(4):963-72
Przyszły kierunek: Szczegółowe badanie chromosomu 11 (duża liczba potencjalnych genów R, interesujące warianty strukturalne) – dla dokładniejszej sekwencji ryżu. Analiza genomu pszenicy przy pomocy znalezionych sekwencji ryżu (ze szczególnym uwzględnieniem sekwencji zawierających NBS) – obecny temat. 3.Uwagi: • potrzeba wzięcia pod uwagę szybko zmiennych, olbrzymich ilości danych – klastrów EST (znaczna część danych) • analiza sekwencji na poziomie DNA (nie białka) • niemożliwe wyszukiwanie pełnych domen (średnia długość EST około 500 nt)
EST fishing: Selekcja 153 sekwencje zawierające NBS, z chromosomów 11 i 12 Oryza sativa ssp. japonica Ekstrakcja charakterystycznych, zakonserwowanych w ewolucji motywów Przeszukiwanie >560,000 ESTs (etykietek ekspresyjnych) pszenicy.
Poszukiwany kandydat - przykład: gi|20112182|gb|BJ300496.1|BJ300496 LENGTH = 672 COMBINED P-VALUE = 1.62e-26 E-VALUE = 8.1e-21 DIAGRAM: 241_[+1b]_177_[+2b]_158 [+1b] P-loop 2.4e-20 V..V..S..I..V..G..M..G..G..L..G..K..T..T..L..A..Q..Q..V..Y. + + + + + + + + + + + + + + + + + + + T..V..S..I..V..G..F..G..G..M..G..K..T..T..L..A..K..A..V..Y. 226 GAAGCATCCATTGAAGACGGTTTCTATTGTTGGATTTGGTGGGATGGGCAAGACAACTCTTGCCAAAGCAGTGTA obszar potencjalnie zmienny (projektowanie primera) .N.. + .D.. 301 TGACAAGCTCAAAGTGCAATTTGATTGTGGTGCCTTTGTTTCAGTTTCTCAAAATCCCGACATCAAGAAGGTTTT [+2b] Kin-2 6.8e-15 K..R..Y..F..I..V..I..D..D..V..W.. + + + + + + + + + + + K..R..Y..L..I..V..I..D..D..I..W.. 451 GATCGATGAAATCATTGAATTTCTTAATGACAAGAGGTATCTCATCGTAATTGATGATATATGGAATGAAAAATC • konserwatywne, dobrze dopasowane wystąpienia motywu • podobieństwo do znanych genów odpornościowych
EST fishing: Selekcja 153 sekwencje zawierające NBS, z chromosomów 11 i 12 Oryza sativa ssp. japonica Ekstrakcja charakterystycznych, zakonserwowanych w ewolucji motywów (program MEME) Przeszukiwanie >560’000 ESTs (etykietek ekspresyjnych) pszenicy. Konstrukcja primerów na podstawie rejonów zmiennych pomiędzy konserwatywnymi motywami.
„Transgeneza i genomika roślin uprawnych” Założona pod koniec 2003 roku Sieć Naukowa KBN. Skupia zespoły naukowe z Poznania, Warszawy, Wrocławia, Radzikowa... Poświęcona promocji wyników badań, koordynacji współpracy między ośrodkami członkowskimi, tworzeniu i upowszechnianiu analiz bioinformatycznych. http://www.cropnet.pl
Serwer sieciowy/ Serwer baz danych Klastr obliczeniowy Końcówka Końcówka Końcówka Końcówka „Transgeneza i genomika roślin uprawnych” http://www.cropnet.pl • porównywanie sekwencji (BLAST, WU-BLAST, CLUSTAL) • wyszukiwanie domen (HMMer, WISE-2.2, PRODIV-TMHMM) • wyszukiwanie zakonserwowanych motywów (MAST/MEME) • rekonstrukcja filogenezy (PHYLIP, NJTREE) • analiza sekwencji (EMBOSS) • predykcja struktury drugorzędowej białek (PSIPRED) • wyszukiwanie powtórzonych sekwencji (RepeatMasker) Aktualny projekt badawczy: analiza zdarzeń insercji/delecji w genomach Arabidopsis thaliana Col-0 i Ler