Download
1 / 25
340 likes | 860 Views
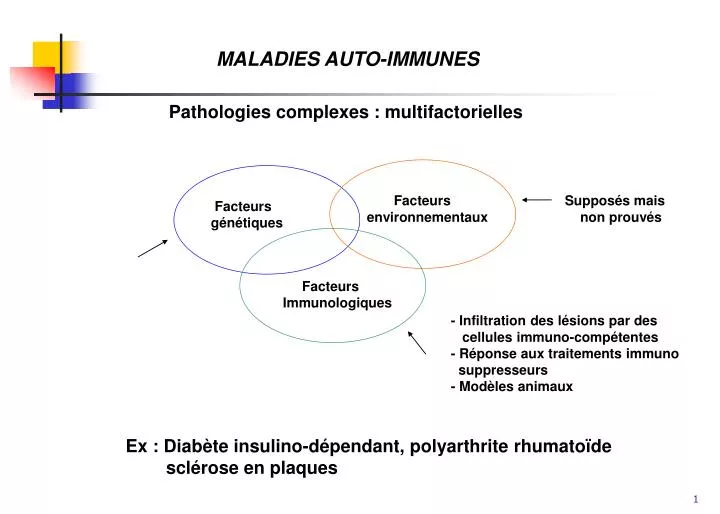
MALADIES AUTO-IMMUNES. Pathologies complexes : multifactorielles. Facteurs environnementaux. Supposés mais non prouvés. Facteurs génétiques. Facteurs Immunologiques. - Infiltration des lésions par des cellules immuno-compétentes - Réponse aux traitements immuno

E N D
MALADIES AUTO-IMMUNES Pathologies complexes : multifactorielles Facteurs environnementaux Supposés mais non prouvés Facteurs génétiques Facteurs Immunologiques - Infiltration des lésions par des cellules immuno-compétentes - Réponse aux traitements immuno suppresseurs - Modèles animaux Ex : Diabète insulino-dépendant, polyarthrite rhumatoïde sclérose en plaques
MALADIES AUTO-IMMUNE Définition : Traduction d’une rupture des mécanismes de la tolérance immunitaire qui contrôlent à l’état physiologique, le niveau d’activation des lymphocytes T et B périphériques vis-à-vis des auto-antigènes exprimés par les tissus de l’organisme Prévalence des MAI dans la population générale : 6 à 7 % (~ 30 MAI répertoriées) Mécanismes contrôlant la tolerance : - délétion clonale - anergie - controle par cellules régulatrices
CLASSIFICATION DES MALADIES AUTO-IMMUNES Historiquement deux groupes principaux : Les maladies auto-immunes spécifiques d’organes : Lésions limitées à un tissu, secondaires à une réaction immunitaire dirigée contre des auto-antigènes dont la distribution est limitée à ce tissu Les maladies auto-immune systémiques : Lésions plus étendues, secondaires à une réaction auto-immune dirigée contre les auto-antigène de distribution ubiquitaire – Ex : le Lupus Pb : une telle identification ne prend pas en compte les mécanismes physiopathologiques D’autres classifications possible en fonction des mécanismes effecteurs impliqués : - MAI à auto-anticorps - MAI à LyT - MAI à dépôts d’immuns complexes
EXISTENCE D’UNE AUTO-IMMUNITE PHYSIOLOGIQUE Des auto-anticorps « naturels » sont présents dans le plasma de sujets sains qui diffèrent des auto-anticorps observés dans les MAI Affinité faible, isotype IgM (le plus souvent ces auto-anticorps n’ont pas été soumis aux mutations somatiques à l’origine de la haute affinité des auto-anticorps de type immuns). Des cellules T auto-réactives existent chez les sujets sains (anti-MBP ou anti-GAD; sans pour autant que ces sujets ne développent une SEP ou un DID) Questions Quels paramètres quantitatifs ou qualitatifs conditionnent la pathogénicité d’un auto-anticorps ou d’une cellule T auto-réactive ? Comment se fait le passage de l’auto-immunité physiologique à l’AI pathologique ? Par quels mécanismes les cellules T pathogènes émergent et se différencient pour Conduire à une MAI ? QUELS SONT LES MECANISMES DE RUPTURE DE LA TOLERANCE AU SOI ?
LA RUPTURE DE LA TOLERANCE Hypothèse : Les cellules auto-réactives émergeraient par activation polyclonale non spécifique des cellules auto-réactives physiologiques Problème : Dans les modèles animaux, dans la majorité des cas, la présence de l’auto-antigène cible est indispensable à l’expression des clones pathogènes Ex : si ablation de l’organe : pas d’auto-anticorps dans la thyroïdite du poulet pas de DID dans le diabète des souris NOD L’activation spécifique des cellules immuno-compétentes par l’auto-antigène cible est donc indispensable. Si absence d’activation l’auto-antigène est ignoré malgré la présence de de cellules potentiellement auto-réactives
AUTO-IMMUNITE CONTRE DES Ag IGNORES Modèle de la souris RIP-GP transgénique Expression de la GP du LCMV sous contrôle du promoteur de l ’insuline de rat néo-antigène exprimé sur les C. du pancréas .Jamais de diabète malgré Ly T auto-réactifs potentiels en périphérie (néo-antigène ignoré) .Si infection par LCMV MAI anti-LCMV avec destruction pancréas Conclusion: Nécessité d ’activer les Ly T circulants pour lever l’ignorance
ACTIVATION DES LYMPHOCYTES T AUTO-REACTIFS Nécessité de deux signaux complémentaires : .Engagement TCR / complexe MHC peptide . Engagement des molécules de co-stimulation (B7.1, B7.2) ligand sur le lymphocyte T : CD28 Conséquence de la nécessité du 2è signal : B7 uniquement représenté sur cellules présentatrices spécialisées Cellules auto-réactives ayant échappé à la sélection négative thymique (Ag absent du thymus) ou cellules de faible ou de moyenne activité : . non activées par l ’antigène exprimé sur les cellules périphériques .anergisées si l ’antigène est rencontré en l ’absence de B7 induction d’une tolérance périphérique Donc nécessité d’une activation des LyT auto-réactifs pour déclencher la MAI
MECANISMES D’ACTIVATION LYMPHOCYTAIRE DANS L’AUTO-IMMUMUNITE TROIS GRAND PROCESSUS : 1 – L’inflammation 2 – Le mimétisme moléculaire ou les réactivites croisées 3 – L’activation polyclonale par des super-antigènes
L’INFLAMMATION Origines diverses de l’inflammation de l’organe cible : . Infection virale . Produit chimique pro-inflammatoire ou toxique Mécanismes en cause : Au cours de l’inflammation, production de cytokines qui amplifient l’expression des molécules promouvant la reconnaissance des auto-antigènes (HLA) Ex : sur-expression par transgénèse de l’INF gamma dans les cellules beta des ilots de Langerhans insulite et diabète
NOTION D’ANTIGENE SPREADING Au cours de la réaction inflammatoire, phénomène de diversification de la réponse auto- immune : . les lésions inflammatoires dévoilent de nouveaux antigènes, différents de l’antigène initial (antigènes cryptiques démasqués) qui donnent lieu au recrutement et à l’activation de nouveaux clones auto-réactifs amplification et chronicité de la réponse pathologique . à terme, difficile de distinguer les premiers antigènes reconnus Exemple : maladie de Theiler : . 1ère phase : le virus infecte les oligodendrocytes . 2è phase : les oligodendrocytes deviennent les cibles d’une réaction T dirigée contre les protéines virales exprimées dans la gaine de la myéline . 3è phase : l’inflammation provoque l’activation des cellules T spécifiques des auto-antigènes de la myéline MAI
MODELE DE LA CROSS-REACTIVITE DU TCR La reconnaissance du TCR n ’est pas unique .un TCR donné peut reconnaître plusieurs ligands avec des affinités différentes .des cellules T de spécificité différente peuvent être sélectionnées par un même complexe HLA peptide dans le thymus certaines d ’entre elles auto-réactives Cette reconnaissance dégénérée du TCR : propriété du répertoire .base de la production thymique de C. pathogéniques .base de l ’auto-immunité par « molecular mimicry »
MODELE DE LA SEP Clones spécifiques de la MBP génèrés chez des patients .à partir de la structure TCR / HLA peptide interrogation de banques de données à la recherche de peptides viraux potentiellement cross-réactifs .réactivité des clones testés contre ces peptides prolifération (herpès simplex, EBV, adenovirus. . . .) Un même TCR capable de reconnaître .5 peptides viraux .1 peptide du Soi auto-antigène candidat dans la SEP
APPROCHE INVERSE Génération de lymphocytes T transgéniques pour le TCR spécifique du LCMV Reconnaissance par ces clones virus spécifiques d ’un antigène des glandes surrénales Infection par le virus LCMV de l ’animal transgénique infiltration de la glande Conclusion :La cross-réactivité entre un auto-antigène et un antigène de l’environnement peut conduire à l’activation de LyT autoréactifs et à l’auto-immunité
ACTIVATION DES LyT AUTO-REACTIFS PAR LES SUPER-ANTIGENES RAPPEL SUR LES SUPER-ANTIGENES Contrairement aux antigènes classiques qui doivent être processés (découpés en peptides) pour être présentés via le CMH aux Ly T et reconnus par un TCR spécifique, les super-antigènes se lient au CMH et au TCR de façon différente qui leur permet de stimuler un grand nombre de lymphocytes T. Les super-antigènes réalisent un pont entre la face externe de la molécule HLA de classe II et la région VB du TCR avec une capacité à lier plusieurs régions VB différentes. Stimulation d’une grande proportion des LyT CD4 (jusqu’à 20 % parmis lesquels des clones auto-réactifs). Ex : Entérotoxines staphylococciques protéines rétro-virales
ROLE DES Ly T REGULATEURS (LTR) : CD4+ CD25+ Arguments en faveur du rôle des LTR .animaux lymphopéniques auto-immunité (gastrite, thyroïdite…) Reconstitution par Ly T CD4 d ’animaux normaux prévention .souris nude reconstituée avec LyT CD4+ déplétés en CD25+ - auto-immunité - si injection CD4+ CD25+ prévention . Injection de LyT CD4+ d ’animaux normaux à souris transgénique avec un TCR MBP spécifique inhibition EAE 1 - des LTR font partie du répertoire T normal 2 - contrôlent les phénomènes d ’auto-immunité 3 - appartiennent à une fraction T CD4+ CD25+ Conclusion : un déficit en LTR peut favoriser l’auto-immunité
APOPTOSE ET AUTO-IMMUNITE : DID Rôle ++ des interactions FAS / FAS-Ldans la cytotoxicité T dépendante, polyclonale non spécifique, non HLA restreint .pas d ’expression de FAS sur les cellules beta .au cours de l ’insulite : - les cellules B expriment FAS sous l ’induction de NO + IL-1 - les cellules B au voisinage des LyT FAS-L + ont un phénotype apoptotique rôle FAS / FAS-L dans la destruction diabétique des îlots .en faveur de cette hypothèse les souris NOD lpr/lpr déficiente en FAS ne développent pas de DID ni spontanément ni après transfert cellules T Mécanismes suggérés : .phase initiale de réponse spécifique contre un nombre limite d ’antigènes .activation type HSR .synthèse de l ’IL-1 et de NO .expression de FAS sur les îlots B .sensibilité à la destruction via LyT-FAS L + SPECIFICITE DE l ’ATTEINTE BETA Conclusion : L’activation anormale des voies apoptotiques peut conduire à la destruction des tissus autologues et à l’auto-immunité
MECANISMES IMMUNOLOGIQUES RESPONSABLES DES LESIONS DANS LES MAI (1) 1 - Auto-Anticorps anti-récepteurs : Effet stimulateur du récepteur reproduisant les actions biologiques du ligand physiologique, Ex : maladie de Basedow : . Auto- anticorps anti-récepteur de la TSH croissance des cellules épithéliales thyroidiennes et sécrétion non controlée des hormones thyroidiennes . Ac TSAb : Thyroid Stimulating Antibody . Ac TGI : Thyroid Grouth Immunoglobulin Effet bloquant sur le récepteur Ex : Maladie coeliaque : . Ac anti-récepteur acetyl-choline 2 – Auto-anticorps anti antigènes solubles : Effet de blocage des fonctions de l’antigène et de modification de ses fonctions Physiologiques Ex : Anémie de Biermer . anticorps anti-facteur intrinsèque, qui lie la vitamine B12 et permet son absorption intestinale
MECANISMES IMMUNOLOGIQUES RESPONSABLES DES LESIONS DANS LES MAI (2) 3 - Auto-Anticorps directement cytotoxiques : Fixation des anticorps sur les tissus ou cellules portant l’antigène cible Activation du Complément jusqu’au complexe lytique Intervention de cellules portant un récepteur pour le Fc des Igl : phénomène ADCC (macrophages, cellules NK) Ex : Anémie hémolytique auto-immune . cytolyse intra-vasculaire (Complément dépendante) ou hépatosplénique (ADCC) des GR
MECANISMES IMMUNOLOGIQUES RESPONSABLES DES LESIONS DANS LES MAI (3) Complexes immuns à auto-anticorps : Les CI peuvent être formés à distance du site où ils vont se déposer (Ex dans la circulation) ou au site même où s’est déposé un auto-antigène Interagissant avec une membrane biologique (Ag planté). Le dépôt de CI modification de la perméabilité des membranes biologiques, activation du Complément, libération des facteurs anaphylactiques et chimiotactiques (C3a, C4a, C5a), recrutement de cellules immuno-compétentes responsables de l’inflammation locale. . PNN libération d’enzymes lysosomiales, de dérivés oxygénés lésions . Activation du complexe lytique du Complément . Cellules possédant un récepteur pour le Fc des Igl ou le Complément (CR1) Ex : le lupus lésions des parois des vaisseaux, synoviales, reins (complexe ADN / anti ADN +++)
MECANISMES IMMUNOLOGIQUES RESPONSABLES DES LESIONS DANS LES MAI (4) Les Ly B doivent bénéficier d’un effet auxiliaire T: L’activation des LyB auto-réactifs est un préalable à la différenciation en Clones B producteurs d’anticorps . L’activation est spécifique de l’auto-antigène avec intervention de Ly CD4 auxiliaires ayant échappé aux mécanismes de tolérance . L’aide des Ly T auxiliaires amplification préférentielle des Ly B ayant augmenté l’affinité de leur BCR après mutation somatique des gènes V NB : les Ly B auto-réactifs ne sont pas délétès mais tolérants
MECANISMES IMMUNOLOGIQUES RESPONSABLES DES LESIONS DANS LES MAI (5) Les Ly T cytotoxiques : Les Ly T CD8 reconnaissant sur le tissu cible les peptides auto-antigéniques présentés par les molécules HLA de classe I (présentes sur la plupart des cellules de l’organisme) Ex : le DID . Prédominance de lymphocytes T CD8+ au sein de l’infiltrat de la glande . Expression de protéines directement impliquées dans les mécanismes de cytotoxicité T (perforine, granzyme) L’hypersensibilté retardée : Dépendante des Ly T CD4 + . Intervention des cytokines pro-inflammatoires (INF gamma, TFN alpha, IL-17) produites par les Ly T auto-réactifs CD4+, TH1 ou TH17 Ex : SEP, DID
MECANISMES IMMUNOLOGIQUES RESPONSABLES DES LESIONS DANS LES MAI (6) Les macrophages : Rôle majeur dans la réaction inflammatoire . Activés par des endotoxines (LPS) ou protéines du choc thermique (HSP) constituants d’agents pathogènes . Activés par des complexes Ag/AC via leur récepteur pour le Fc des immuno-globulines Exemple : la polyarthrite rhumatoïde . Macrophages activés par IC (FR ou Ac anti-CCP) Conséquences : Production de cytokines pro-inflammatoires (IL-1, IL-12, IL-18, TNF) activation des synoviocytes in situ sécrétion d’IL-1 + TNF destruction du cartilage + activation autocrine des synoviocytes production métaloproteases, NO, dérivés oxygènés Production d’IL-6 syndrome inflammatoire général ( CRP, VS, fibrinogène) et prolifération des ostéoclastes à partir des phagocytes mononucléés sécrétion cathepsine K et destruction collagène osseux Production de chimiokines afflux de polynucléaires, monocytes et lymphocytes






















