Download

1 / 44
500 likes | 1.18k Views
SEQUENCAGE DES GENOMES EUCARYOTES (et procaryotes). Séquençage d’ADN • 2 méthodes publiées in 1977 – méthode chimique: Maxam, A.M. and Gilbert, W. (1977) A new method for sequencing DNA. Proc. Natl. Acad. Sci. USA , 74 , 560-564. – méthode biochimique: Sanger, F., Micklen, S.,

E N D
SEQUENCAGE DES GENOMES EUCARYOTES (et procaryotes)
Séquençage d’ADN • 2 méthodes publiées in 1977 – méthode chimique: Maxam, A.M. and Gilbert, W. (1977) A new method for sequencing DNA. Proc. Natl. Acad. Sci. USA, 74, 560-564. – méthode biochimique: Sanger, F., Micklen, S., and Coulson, A.R. (1977) DNA sequencing and chain terminating inhibitors. Proc. Natl. Acad. Sci. USA, 74, 5463-5467.
Séquençage de Maxam-Gilbert Clivage chimique d’ADN marqué à son extrémité 1. Marquage radioactif des extrémités (5' or 3') , 2. Dénaturation de l’ADN 3. Quatre réactions chimiques spécifiques, représentant 4 combinaisons possibles: – G seulement: DMS, piperidine – A + G: DMS, acide formique, piperidine – C+T: Hydrazine, piperidine – C seulement: Hydrazine dans 1.5M NaCl, piperidine
Séquençage de Maxam-Gilbert – le premier composé chimique casse la liaison glycosidique entre le ribose et la base, déplaçant la base. – le traitement piperidine catalyse la coupure de la liaison phosphodiester d’où la base a été déplacée. – les produits de réactions sont soumis à une électrophorèse sur un gel de polyacrylamide en condition dénaturante. Les fragments les plus petits se déplacent le plus facilement. La séquence est lue du bas du gel (5’) vers le haut du gel (3’).
Séquençage de Maxam-Gilbert • le principal avantage de cette technique est qu’elle n’est pas dépendante des problèmes de synthèse d’ADN par une polymérase (terminaison précoce due à la séquence ou à la structure de l’ADN). • le principal inconvénient est la toxicité des composés chimiques utilisés.
Technique de séquençage de SANGER • méthode biochimique • aussi appelée séquençage par terminaison de chaîne • ou aux dideoxy. • basée sur l’incorporation d’un dideoxynucléotide à l’extrémité • d’une molécule d’ADN en cours de synthèse.
Technique de séquençage de SANGER 1- hybridation du primer de séquençage sur la matrice simple brin à séquencer. 2- préparation des 4 mélanges réactionnels en parallèle. Chaque mélange contient chacun des 4 dNTP (un est marqué en α avec du 32P, du 35S ou du 33P) et un des 4 ddNTP. 3- la réaction démarre lorsque la DNA polymérase est ajoutée au mélange (Klenow, T7, Taq)
Technique de séquençage de SANGER 4- la synthèse du brin d’ADN cesse par l’incorporation d’un ddNTP et la réaction est arrêtée par l’addition du tampon de charge du gel de séquençage contenant de la formamide. 5- chauffage des échantillons pour défaire les structures de l’ADN avant de charger sur le gel dénaturant de polyacrylamide/urée pré-chauffé. 6- les petits fragments migrent plus loin. L’extremité 5’ est en bas du gel et l’extrémité 3’ en haut. 7- la séquence lue est la séquence complémentaire de la matrice.
Technique de séquençage de SANGER La séquence de la matrice est la séquence complémentaire de la séquence lue sur le gel.
Séquençage d’ADN automatisé • Version améliorée de la méthode de Sanger: • marquage radioactif marquage fluorescent des ddNTP • film autoradiographique détection par faisceau laser • en cours d’électrophorèse • polymérase de Klenow Taq polymérase • quantité de matrice quantité plus faible que pour la méthode • de Sanger classique car thermocyclage
Séquençage d’ADN automatisé • procédure de séquençage basique en cycle • - hybridation du primer sur la matrice sous forme simple brin • - extension du primer lors d’une réaction limitante en • ddNTP fluorescent et en excès de dNTP (rapport 1/100). • - dénaturation et redémarrage d’un nouveu cycle • détection par émission de fluorescence après stimulation du colorant • fluorescent; couleur et position sont enregistrée dans un fichier séquence. • format de sortie du fichier: chromatogramme ou fichier de séquence
Le séquençage des génomes Les choix stratégiques Approches utilisées pour le séquençage à grande échelle Organismes séquencés Identification des gènes Génomes procaryotes Structure chromosomique Organisation des gènes Séquences non codantes Retombées médicales et commerciales Génomes des modèles eucaryotes Structure des chromosomes Identification des gènes Fonctions des gènes reconnus ou prédits Régions non codantes Génome humain Les chromosomes humains Identification des gènes Séquences répétées
Le séquençage des génomes Les choix stratégiques Approches utilisées pour le séquençage à grande échelle Organismes séquencés Identification des gènes Génomes procaryotes Structure chromosomique Organisation des gènes Séquences non codantes Retombées médicales et commerciales Génomes des modèles eucaryotes Structure des chromosomes Identification des gènes Fonctions des gènes reconnus ou prédits Régions non codantes Génome humain Les chromosomes humains Identification des gènes Séquences répétées
Les choix stratégiques Approches utilisées pour le séquençage à grande échelle Deux approches : Multitude de laboratoires : 46 laboratoires pour B subtilis en 1997 34 laboratoires pour Xylella fastidiosa en 2000 35 laboratoires pour la levure en 1991 Genome Centers : Grande échelle de production Séquenceurs automatiques Organismes séquencés • Recherche fondamentale : E coli, B subtilis, S. pombe, A thaliana, drosophile, nématode, Neurospora crassa • Utilisation industrielle : Agrobacterium tumefaciens, Lactococcus lactis, Archébactéries (haute température, métabolismes particuliers) • Intérêt médical : procaryotes pathogènes
Stratégies de séquençage des génomes complets méthode dite « bac-to-bac » ou « map-based » méthode dite de « shotgun » L’approche « bac-to-bac » passe par la création d’une carte physique brute de l’ensemble du génome avant le séquençage. La construction de la carte nécessite de couper les chromosomes en grands fragments et de déterminer la position relative de ces fragments avant de les séquencer. La méthode de “shotgun” passe directement par l’étape de séquençage Sans création d’une carte physique (évidement ça paraît plus facile).
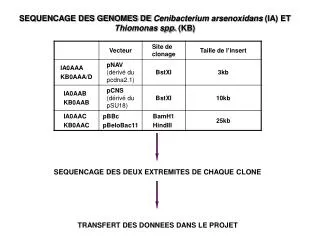
1- plusieurs copies du génome sont coupées au hasard en fragments d’environ 150 kpb. Stratégies de séquençage des génomes complets Les étapes BAC to BAC SHOTGUN 1- plusieurs copies du génome sont cassées au hasard en fragments de 2 kpb en faisant passer l’AND sous pression dans l’aiguille d’une seringue. Cette étape est renouvelée de façon à générer des fragments de 10 kpb. 2- chacun des fragments est inséré dans un BAC constituant ainsi la banque BAC. 2- chaque fragment de 2 ou 10 kpb est inséré dans un plasmide.
3- chaque fragment est marqué d’une empreinte qui va donné à chaque BAC une identification Unique qui va permettre de déterminer l’ordre des fragments les uns par rapport aux autres. L’empreinte est obtenue en coupant chaque fragment du BAC par un enzyme et en séquençant l’extrémité du BAC afin de positionner les BAC le long des chromosomes. 3- chaque banque de plasmides de 2 ou 10 kpb est séquencée. 500 pb de l’extrémité de chaque fragment sont décodées. Le séquençage de chacune des extrémités est déterminant pour l’assemblage de l’ensemble des chromosomes. 4- des algorithmes assemblent les millions de fragments séquencés en un ensemble continu correspondant à chaque chromosome. 4- Chaque BAC est cassé au hasard en fragments d’environ 1, 5 kpb clonés dans des phagemides.
5- chaque banque de phage est séquencée. 500 pb de l’extrémité de chaque fragment sont séquencées. 6- ces séquences alimentent un programme informatique appelé PHRAP qui identifie les séquences communes qui joignent 2 fragments adjacents.
Comparaison des cartes du génomes d’Arabidopsis thaliana séquence génétique physique
Les choix stratégiques Identification des gènes • Identification facile chez les Procaryotes : • promoteurs, séquences codantes, signaux de terminaison • Pas ou peu de séquences intergéniques • Identification difficile chez les Eucaryotes : • Découpage des gènes en introns et exons • Régions intergéniques parfois très vastes Levure : 5% des gènes sont morcelés et régions non-codantes peu abondantes Nématode, Drosophile, Arabette : régions codantes majoritairement fragmentées et régions non-codantes très étendues • Comparaison des séquences génomiques et des séquences d’ADNc (EST ou séquence complète d’ARNm)alignement : séquence transcrite • Outils informatiques de prédiction : recherche de phase ouverte de lecture, signaux d’épissage, composition en bases • Utilisation des données d’un autre organisme. Ex : EST de Caenorhabditis briggsae pour Caenorhabditis elegans
Le séquençage des génomes Les choix stratégiques Approches utilisées pour le séquençage à grande échelle Organismes séquencés Identification des gènes Génomes procaryotes Structure chromosomique Organisation des gènes Séquences non codantes Retombées médicales et commerciales Génomes des modèles eucaryotes Structure des chromosomes Identification des gènes Fonctions des gènes reconnus ou prédits Régions non codantes Génome humain Les chromosomes humains Identification des gènes Séquences répétées
Génomes procaryotes Structure chromosomique Abondance en guanine et cytosine Un faible taux de G+C indique souvent un mode de vie parasitique ou synbiotique La réplication du chromosome se fait dans deux directions opposées divergeant à partir de l’origine de réplication. Chacune de ces deux moitiés est appelée réplichore Le séquençage révèle parfois des plasmides, des plasmides linéaires ou des mégaplasmides
Génomes procaryotes Organisation des gènes • La fraction codante est élevée (environ 90%) • La taille moyenne des gènes est de 1 kb • Le nombre de gènes est très variable (500 à 8000) • Les unités transcriptomiques sont fréquemment organisées en opérons • Les gènes codant pour les ARNr sont le plus souvent agencés en 16S-23S-5S avec des gènes d’ARNt entre les gènes • Le nombre de pseudogènes (gènes mutés non-transcrits ou non-traduits) est faible. • Exception Mycobacterium leprae avec 24% de régions non codantes et 27% de gènes.
Génomes procaryotes Séquences non codantes • Régions intergéniques (séquences régulatrices, parfois des séquences répétées et quelques rares introns) • Chez E coli taille moyenne des régions intergéniques :118 pb • Les séquences répétées en tandem comprennent un motif de 1 à 6 nt répété de 2 à quelque dizaine de fois • Les séquences dédiées à la transformation comme les USS (Uptake Signal Sequence) de H influenzae (1465 USS par génome)
Génomes procaryotes Retombées médicales et commerciales • De nombreuses retombées médicales sont espérées : • La syphilis touche 50 106 de personnes • La lèpre touche 15 106 de personnes • Chaque minute la tuberculose atteint 10 personnes • La comparaison de génomes d’espèces proches mais causant des maladies très différentes comme Mycobacterium leprae , Mycobacterium tuberculosis , Neisseria meningitidis , devrait permettre d’identifier les gènes responsables de tel ou tel autre effet pathogène • Diagnostic ou pronostic de développement d’infection (ex recherche de la séquence répétée Ng-rep utilisée pour détecter une contamination par Neisseria meningitidis ) • Des protéines de bactéries extrêmophiles sont commercialisées (ex la Taq de Thermus aquaticus)
Le séquençage des génomes Les choix stratégiques Approches utilisées pour le séquençage à grande échelle Organismes séquencés Identification des gènes Génomes procaryotes Structure chromosomique Organisation des gènes Séquences non codantes Retombées médicales et commerciales Génomes des modèles eucaryotes Structure des chromosomes Identification des gènes Fonctions des gènes reconnus ou prédits Régions non codantes Génome humain Les chromosomes humains Identification des gènes Séquences répétées
Génomes des modèles eucaryotes Structure des chromosomes • Chez la levure , les régions riches en G+C correspondent aux régions riches en gènes. Les brins complémentaires codent pour un nombre similaire de gènes sauf pour le chromosome II et pour la région centrale du chromosome VI • Chez C elegans le génome est remarquablement uniforme en teneur G+C le long des chromosomes. La densité des gènes est plus élevées dans les régions centrales que dans les bras chromosomiques. La densité des gènes est faible sur le chromosome X. • Chez la drosophile, 180 Mb avec 60 Mb d’hétérochromatine (séquence répétée, éléments transposables, deux blocs de gènes ribosomiques). L’euchromatine couvre 120 Mb qui contient la majorité des gènes. • Chez la souris 20 paires de chromosomes (19 autosomes et une paire de chromosomes sexuels) tous acrocentriques. • Chez A thaliana, 5 chromosomes tous autosomiques (2 acrocentriques, 2 submétacentriques et 1 métacentrique. L'hétérochromatine ne change pas d'état de condensation au cours du cycle cellulaire si le bras court est presque aussi long que le bras long, le chromosome est dit métacentrique; s'il est plus court, il est dit sub-métacentrique. Enfin, si ce bras p est très petit, le chromosome est dit acrocentrique
Génomes des modèles eucaryotes Identification des gènes
Génomes des modèles eucaryotes Fonctions des gènes reconnus ou prédits • Prédiction de fonction : le nombre de gènes potentiellement impliqués dans une fonction biologique donnée s’est soudainement accru avec le séquençage systématique (selon l’espèce 40 à 60 % des gènes ne sont toujours pas reliés à des gènes de fonction connue) • Chez la levure : identification d’un nouveau gène codant pour l’histone H1. • Chez le nématode : identification de protéines SXC impliquées dans des interactions avec la matrice extracellulaire. • Chez l’Arabette : identification d’un gène codant pour la lyase hydroxynitrile qui produit de l’acide cyanhydrique (répulsif d’herbivores) • Les gènes codant pour les cyclines de la levure sont différents de ceux très similaires de la drosophile, du nématode, des vertébrés
Génomes des modèles eucaryotes Régions non codantes Plus faible que chez l’homme Séquences répétées en tandem : les microsatellites : répétitions de motifs de 1 à 13 nt, polymorphes et distribués le long des chromosomes Les minisatellites : répétitions de motifs de 14 à 500 nt, distribués sur 0,5 à 30 kb. Séquences répétées dispersées : (40 % du génome murin) LINE, SINE, rétrotransposons à LTR et les rétrotransposons à ADN
Microsatellite Minisatellite
Le séquençage des génomes Les choix stratégiques Approches utilisées pour le séquençage à grande échelle Organismes séquencés Identification des gènes Génomes procaryotes Structure chromosomique Organisation des gènes Séquences non codantes Retombées médicales et commerciales Génomes des modèles eucaryotes Structure des chromosomes Identification des gènes Fonctions des gènes reconnus ou prédits Régions non codantes Génome humain Les chromosomes humains Identification des gènes Séquences répétées
Génome humain Les chromosomes humains La longueur totale du génome humain : 3000 Mb 20 laboratoires de 6 pays (USA, GB, Japon, France, Allemagne et Chine) 1000 nt / sec
Génome humain Identification des gènes • 535 gènes codant pour des ARNt (plus faible que chez le nématode et plus élevé que chez la drosophile) • 150 à 200 groupes de gènes codant pour les ARNr 18S, 28S et 5,8S sur les chromosomes 13, 14, 15, 21 et 22 • 2000 gènes codant pour l’ARNr 5S sur le chromosome 1 • Les gènes codants pour des protéines ont été prédits : • Comparaison aux bases de données d’EST • Comparaison aux séquences complètes d’ARNm • Programme de prédiction comme GENESCAN • Le nombre total de gènes varient entre 26000 et 35000 gènes (2x plus que le nématode ou la drosophile). • 11,1 gènes / Mb • Taille moyenne des gènes 27900 nt répartis en 8 à 9 exons de 145 nt environ avec des introns d’environ 3500 nt. Plus de 35 % des gènes ont un épissage alternatif • 28% du génome serait transcrit en ARNr, ARNm, ARNt ou ARN de petite taille et 1,4 % serait traduit. • Le gène le plus grand est celui de la dystrophine (2,4 Mb) • Le plus grand messager est celui de la titine (80780 nt) avec 178 exons et l’exon le plus grand (17 106 nt)
Génome humain Séquences répétées