Download
1 / 1
10 likes | 150 Views
Mitochondrial oxidative damage-induced cellular senescence in mouse keratinocytes is associated with altered wound healing and epidermal stem cell depletion during aging * Michael C. Velarde 1 , Simon Melov 1 , Judith Campisi 1,2 1 Buck Institute for Age Research, Novato, CA, USA, 94945

E N D
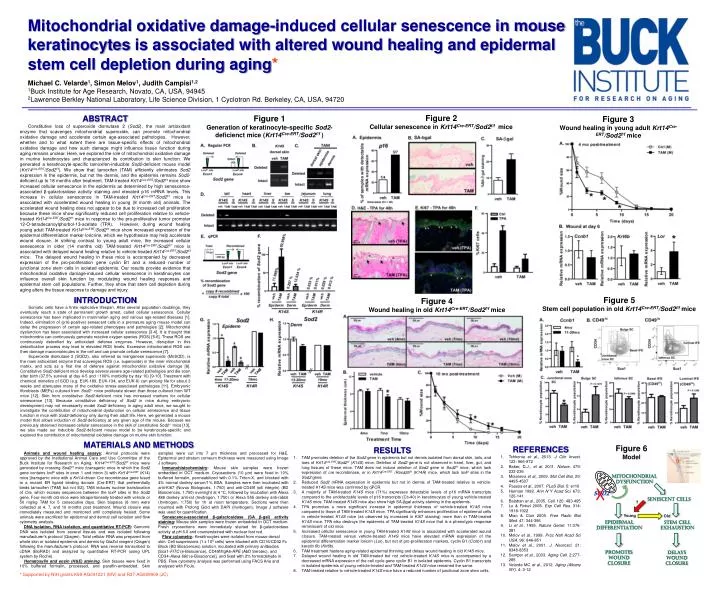
Mitochondrial oxidative damage-induced cellular senescence in mouse keratinocytes is associated with altered wound healing and epidermal stem cell depletion during aging* Michael C. Velarde1, Simon Melov1, Judith Campisi1,2 1Buck Institute for Age Research, Novato, CA, USA, 94945 2Lawrence Berkley National Laboratory, Life Science Division, 1 Cyclotron Rd. Berkeley, CA, USA, 94720 Figure 2 Cellular senescence in Krt14Cre-ERT/Sod2f/f mice ABSTRACT Constitutive loss of superoxide dismutase 2 (Sod2), the main antioxidant enzyme that scavenges mitochondrial superoxide, can promote mitochondrial oxidative damage and accelerate certain age-associated pathologies. However, whether and to what extent there are tissue-specific effects of mitochondrial oxidative damage and how such damage might influence tissue function during aging remains unclear. Here, we explored the role of mitochondrial oxidative damage in murinekeratinocytes and characterized its contribution to skin function. We generated a keratinocyte-specific tamoxifen-inducible Sod2-deficient mouse model (Krt14Cre-ERT/Sod2f/f). We show that tamoxifen (TAM) efficiently eliminates Sod2 expression in the epidermis, but not the dermis, and the epidermis remains Sod2-deficient up to 18 months after treatment. TAM-treated Krt14Cre-ERT/Sod2f/f mice show increased cellular senescence in the epidermis as determined by high senescence-associated β-galactosidase activity staining and elevated p16 mRNA levels. This increase in cellular senescence in TAM-treated Krt14Cre-ERT/Sod2f/f mice is associated with accelerated wound healing in young (8 month old) animals. The accelerated wound healing does not appear to be due to increased cell proliferation because these mice show significantly reduced cell proliferation relative to vehicle-treated Krt14Cre-ERT/Sod2f/f mice in response to the pro-proliferative tumor promoter 12-O-tetradecanoylphorbol-13-acetate (TPA). However, during wound healing young adult TAM-treated Krt14Cre-ERT/Sod2f/f mice show increased expression of the epidermal differentiation marker loricrine, which we hypothesize may help accelerate wound closure. In striking contrast to young adult mice, the increased cellular senescence in older (14 months old) TAM-treated Krt14Cre-ERT/Sod2f/f mice is associated with delayed wound healing relative to vehicle-treated Krt14Cre-ERT/Sod2f/f mice. The delayed wound healing in these mice is accompanied by decreased expression of the pro-proliferation gene cyclinB1 and a reduced number of junctional zone stem cells in isolated epidermis. Our results provide evidence that mitochondrial oxidative damage-induced cellular senescence in keratinocytes can influence overall skin function bymodulating wound healing responses and epidermal stem cell populations. Further, they show that stem cell depletion during aging alters the tissue response to damage and injury. Figure 1 Generation of keratinocyte-specific Sod2-deficienct mice (Krt14Cre-ERT/Sod2f/f ) Figure 3 Wound healing in young adult Krt14Cre-ERT/Sod2f/f mice INTRODUCTION Somatic cells have a finite replicative lifespan. After several population doublings, they eventually reach a state of permanent growth arrest, called cellular senescence. Cellular senescence has been implicated in mammalian aging and various age-related diseases [1]. Indeed, elimination of (p16-positive) senescent cells in a premature aging mouse model can delay the progression of certain age-related phenotypes and pathologies [2]. Mitochondrial dysfunction has been associated with increased cellular senescence [3-4]. It is thought that mitochondria can continuously generate reactive oxygen species (ROS) [5-6]. These ROS are continuously detoxified by antioxidant defense enzymes. However, disruption in this detoxification process may lead to elevated ROS levels. Excessive mitochondrial ROS can then damage macromolecules in the cell and can promote cellular senescence [7]. Superoxide dismutase 2 (SOD2), also referred as manganese superoxide (MnSOD), is the main antioxidant enzyme that scavenges ROS (i.e. superoxide) in the inner mitochondrial matrix, and acts as a first line of defense against mitochondrial oxidative damage [8]. Constitutive Sod2-deficient mice develop several severe age-related pathologies and die soon after birth (37.5% survival at day 4-5 and ~100% morbidity by day 10) [9-10]. Treatment with chemical mimetics of SOD (e.g. EUK-189, EUK-134, and EUK-8) can prolong life for about 3 weeks and attenuates many of the oxidative stress-associated pathologies [11]. Embryonic fibroblasts (MEFs) cultured from Sod2-/- mice proliferate slower than those cultured from WT mice [12]. Skin from constitutive Sod2-deficient micehas increased markers for cellular senescence [13]. Because constitutive deficiency of Sod2 in mice during embryonic development may not necessarily model Sod2 deficiency in aging adult mice, we sought to investigate the contribution of mitochondrial dysfunction on cellular senescence and tissue function in mice with Sod2-deficiency only during their adult life. Here, we generated a mouse model that allows induction of Sod2-deficiency at any given age of the mouse. Because we previously observed increased cellular senescence in the skin of constitutive Sod2-/- mice [13], we also made our inducible Sod2-deficient mouse model to be keratinocyte-specific and explored the contribution of mitochondrial oxidative damage on murine skin function. Figure 5 Stem cell population in old Krt14Cre-ERT/Sod2f/f mice Figure 4 Wound healing in old Krt14Cre-ERT/Sod2f/f mice MATERIALS AND METHODS Figure 6 Model • REFERENCES • Tchkoniaet al., 2013. J Clin Invest. 123: 966-972 • Baker, D.J., et al. 2011. Nature. 479: 232-236 • Moiseevaet al., 2009. Mol Cell Biol. 29: 4495-4507 • Passoset al., 2007. PLoS Biol. 5: e110 • Harman 1992. Ann N Y Acad Sci. 673: 126-141 • Balabanet al., 2005. Cell. 120: 483-495 • Lu & Finkel 2008. Exp Cell Res. 314: 1918-1922 • Miao & Clair 2009. Free RadicBiol Med. 47: 344-356 • Li et al., 1995. Nature Genet. 11:376-381 • Melov et al., 1999. Proc NatlAcadSci USA. 96: 846-851 • Melov et al., 2001. J. Neurosci. 21: 8348-8353 • Samper et al., 2003. Aging Cell. 2:277-285 • Velarde MC et al., 2012. Aging (Albany NY). 4: 3-12 • RESULTS • TAM promotes deletion of the Sod2 gene in epidermis but not dermis isolated from dorsal skin, tails, and toes of Krt14Cre-ERT/Sod2f/f(K14S)mice. Deletion of Sod2 gene is not observed in heart, liver, gut, and lung tissues of these mice. TAM does not induce deletion of Sod2 gene in Sod2f/fmice, which lack expression of crerecombinase, or in Krt14Cre-ERT /Rosa26f/f (K14R) mice, which lack loxP sites in the Sod2 gene. • Reduced Sod2 mRNA expression in epidermis but not in dermis of TAM-treated relative to vehicle-treated K14S mice was confirmed by qPCR. • A majority of TAM-treated K14S mice (71%) expresses detectable levels of p16 mRNA transcripts compared to the undetectable levels of p16 transcripts (Ct>40) in keratinocytes of young vehicle-treated K14S mice. TAM-treated K14S mice also show high SA-βgal activity staining in the epidermis. • TPA promotes a more significant increase in epidermal thickness of vehicle-treated K14S mice compared to those of TAM-treated K14S mice. TPA significantly enhances proliferation of epidermal cells in vehicle-treated K14S mice (as observed by increased in Ki67 staining) more than in TAM-treated K14S mice. TPA also destroys the epidermis of TAM-treated K14S mice that is a phenotypic response reminiscent of old mice. • Increased cellular senescence in young TAM-treated K14S mice is associated with accelerated wound closure. TAM-treated versus vehicle-treated K14S mice have elevated mRNA expression of the epidermal differentiation marker loricrin (Lor), but not of pro-proliferation markers, cyclinB1 (Ccnb1) and keratin 6b (Krt6b). • TAM treatment hastens aging-related epidermal thinning and delays wound healing in old K14S mice. • Delayed wound healing in old TAM-treated but not vehicle-treated K14S mice is accompanied by a decreased mRNA expression of the cell cycle gene cyclin B1 in isolated epidermis. Cyclin B1 transcripts in isolated epidermis of young vehicle-treated and TAM-treated K14S mice remained the same. • TAM-treated relative to vehicle-treated K14S mice have a reduced number of junctional zone stem cells. Animals and wound healing assays: Animal protocols were approved by the Institutional Animal Care and Use Committee of the Buck Institute for Research on Aging. Krt14Cre-ERT/Sod2f/f mice were generated by crossing Sod2f/f mice (transgenic mice in which the Sod2 gene contains loxP sites in exon 1 and intron 3) with Krt14Cre-ERT (K14) mice [transgenic mice with a Krt14-driven Crerecombinase gene fused to a mutant ER ligand binding domain (Cre-ERT) that preferentially binds tamoxifen (TAM) but not E2]. TAM induces nuclear translocation of Cre, which excises sequences between the loxP sites in the Sod2 gene. Four-month old mice were intraperitoneally treated with vehicle or 50 mg/kg TAM for 5 consecutive days. Skin biopsies (8 mm) were collected at 4, 7, and 10 months post treatment. Wound closure was immediately measured and monitored until completely healed. Some animals were sacrificed for epidermal and dermal cell isolation and flow cytometry analysis. DNA isolation, RNA isolation, and quantitative RT-PCR: Genomic DNA was isolated from several tissues and was isolated following manufacturer’s protocol (Qiagen). Total cellular RNA was prepared from whole skin or isolated epidermis and dermis by QiaZol reagent (Qiagen) following the manufacturer’s protocol. RNA was reverse transcribed to cDNA (BioRAD) and analyzed by quantitative RT-PCR (using UPL system by Roche). Hematoxylin and eosin (H&E) staining. Skin tissues were fixed in 10% buffered formalin, processed, and paraffin-embedded. Skin samples were cut into 7 µm thickness and processed for H&E. Epidermal and stratum corneum thickness were measured using Image J software. Immunohistochemistry: Mouse skin samples were frozen embedded in OCT medium. Cryosections (10 µm) were fixed in 10% buffered formalin, permeabilized with 0.1% Triton-X, and blocked with 4% normal donkey serum/1% BSA. Samples were then incubated with anti-Ki67 (Novus Biologicals, 1:750) and anti-CD49f (α6 integrin) (BD Biosciences, 1:750) overnight at 4 °C, followed by incubation with Alexa 488 donkey anti-rat (Invitrogen, 1:750) or Alexa 555 donkey anti-rabbit (Invitrogen, 1:750) for 1h at room temperature. Sections were then mounted with Prolong Gold with DAPI (Invitrogen). Image J software was used for quantification. Senescence-associated β-galactosidase (SA β-gal) activity staining: Mouse skin samples were frozen embedded in OCT medium. Fresh cryosections were immediately stained for β-galactosidase activity at pH 6.0 and counterstained with nuclear fast red. Flow cytometry: Keratinocytes were isolated from mouse dorsal skin. Cell suspensions (1 x 106 cells) were blocked with CD16/CD32 Fc Block (BD Biosciences) solution, incubated with primary anitbodies [Sca1-FITC (e-Bioscience), CD49f/ItgA6-RPE (AbDSerotec), and CD34-Alexa 660 (e-Bioscience)], and fixed with 2% formaldehyde in PBS. Flow cytometry analysis was performed using FACS Aria and analyzed with FloJo. Mitochondrial Dysfunction X SOD2 Senescent cells ? Young Old EPIDERMAL DIFFERENTIATION Stem cell exhaustion Promotes Wound closure Delays wound closure * Supported by NIH grants K99-AG041221 (MV) and R37-AG009909 (JC)