Download

1 / 26
270 likes | 882 Views
Nobel Laureates of X Ray Crystallography. Max von Laue - 1914 Nobel Prize in Physics Bragg(s) - 1915 Nobel Prize in Physics. Dorothy Hodgkin – 1964 Nobel Prize in Chemistry Hauptman and Karle - 1985 Nobel Prize in Chemistry Roderick MacKinnon and Peter Agre – 2003 Nobel Prize in Chemistry.

E N D
Nobel Laureates of X Ray Crystallography • Max von Laue - 1914 Nobel Prize in Physics • Bragg(s) - 1915 Nobel Prize in Physics. • Dorothy Hodgkin – 1964 Nobel Prize in Chemistry • Hauptman and Karle - 1985Nobel Prize in Chemistry • Roderick MacKinnon and Peter Agre – 2003 Nobel Prize in Chemistry
ψ φ Cα Cα
Levinthal's paradox • How many backbone conformations of a 300 residue protein are possible? • Only taking into account Phi and Psi angles, approximately 10³°° conformations • How is the right conformation found in our lifetime? • Answer: Only some angle combinations are energetically favorable, hence only a limited amount of structural conformations are possible
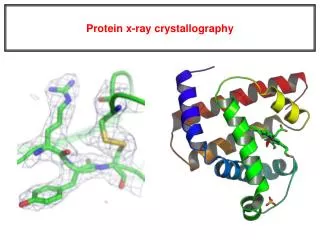
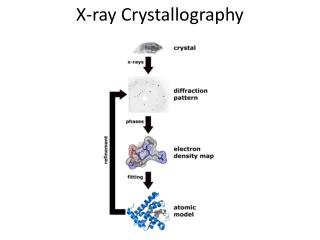
When X-rays strike a macromolecular crystal, the atoms in the molecules produce scattered X-ray waves which combine to give a complex diffraction pattern consisting of waves of different amplitudes
What is measured experimentally are the amplitudes and positions of the scattered X-ray waves from the crystal • X-ray crystallography provides the positions of all non-hydrogen atoms • The origin of each wave must be determined so that they sum to give an image instead of a “sea of noise” • Phase values must be assigned to all of the recorded data; sometimes done computationally, but is usually done experimentally by labeling the protein with one or more heavy atoms whose position in the crystal can be determined independently
Electron Density Calculation • Diffraction amplitudes = FT{Electron density} • Take the inverse FT of the diffraction pattern to regenerate the electron density • Shooting a crystal with X-rays and obtaining a diffraction pattern is the same as taking the Fourier transform of a compound. Hence, taking the Fourier transform again gives us the original structure.
After shooting Our Protein with X-rays And getting the FT Taking the FT of the FT RESTORES the original Data (mostly) Our Protein A very simple example of Fourier and Inverse Fourier transforms
The scattered x-rays have amplitudes given by Fourier coefficients of electron density • Possible to measure amplitudes • If we could also measure phases, we could compute electron density by inverse Fourier transform • We then fit a model to the density • Phases are extremely difficult to measure
Quick Recap • Crystallize Protein (if humanly possible) • Measuring diffraction amplitudes • Using a computer to calculate electron density • Building a model consistent w/ density
Quality Of a Structure • R-factors represent the percentage disagreement between the observed diffraction pattern and that calculated from the final model • R-factors of around 20% or less are considered well determined structures that are expected to contain relatively few errors
Express the resolution of a structure in terms of a distance
Molecular Replacement • Use an analogous structure (similar amino acid sequence) and apply to the structure you are trying to determine • “Replacement” actually means 'relocation, repositioning‘ atoms.