Download
1 / 2
20 likes | 128 Views
Universität Bielefeld SFB 613. K6. Photoinduzierte Dissoziations- und Reaktionsprozesse von Einzelmolekülen in spezifischen organischen Umgebungen unter Berücksichtigung quantenchemischer Rechnungen. R. Brodbeck 1 , V. Koch 1 , Ch. Evenhuis 1 , A. Rozhenko 1 , U. Manthe 1 ;

E N D
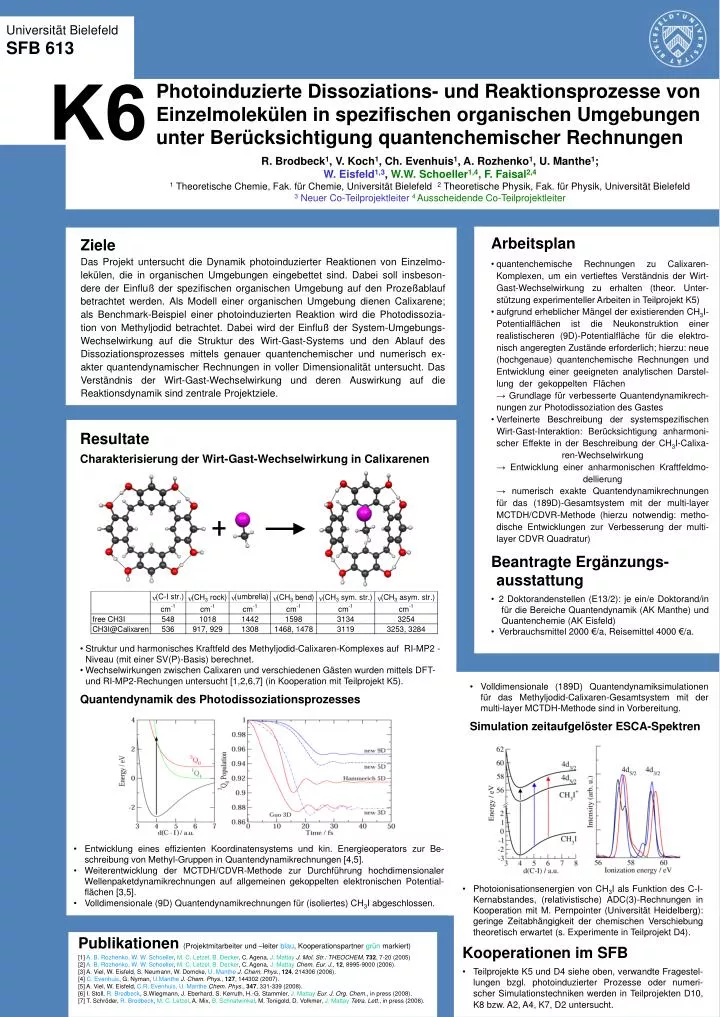
Universität Bielefeld SFB 613 K6 Photoinduzierte Dissoziations- und Reaktionsprozesse von Einzelmolekülen in spezifischen organischen Umgebungen unter Berücksichtigung quantenchemischer Rechnungen R. Brodbeck1, V. Koch1, Ch. Evenhuis1, A. Rozhenko1, U. Manthe1; W. Eisfeld1,3, W.W. Schoeller1,4, F. Faisal2,4 1 Theoretische Chemie, Fak. für Chemie, Universität Bielefeld 2 Theoretische Physik, Fak. für Physik, Universität Bielefeld 3 Neuer Co-Teilprojektleiter4 Ausscheidende Co-Teilprojektleiter • Arbeitsplan • quantenchemische Rechnungen zu Calixaren-Komplexen, um ein vertieftes Verständnis der Wirt-Gast-Wechselwirkung zu erhalten (theor. Unter-stützung experimenteller Arbeiten in Teilprojekt K5) • aufgrund erheblicher Mängel der existierenden CH3I-Potentialflächen ist die Neukonstruktion einer realistischeren (9D)-Potentialfläche für die elektro-nisch angeregten Zustände erforderlich; hierzu: neue (hochgenaue) quantenchemische Rechnungen und Entwicklung einer geeigneten analytischen Darstel-lung der gekoppelten Flächen :→ Grundlage für verbesserte Quantendynamikrech-nungen zur Photodissoziation des Gastes • Verfeinerte Beschreibung der systemspezifischen Wirt-Gast-Interaktion: Berücksichtigung anharmoni-scher Effekte in der Beschreibung der CH3I-Calixa-ren-Wechselwirkung→ Entwicklung einer anharmonischen Kraftfeldmo-dellierung→ numerisch exakte Quantendynamikrechnungen für das (189D)-Gesamtsystem mit der multi-layer MCTDH/CDVR-Methode (hierzu notwendig: metho-dische Entwicklungen zur Verbesserung der multi-layer CDVR Quadratur) • Beantragte Ergänzungs-ausstattung • 2 Doktorandenstellen (E13/2): je ein/e Doktorand/in für die Bereiche Quantendynamik (AK Manthe) und Quantenchemie (AK Eisfeld) • Verbrauchsmittel 2000 €/a, Reisemittel 4000 €/a. Ziele Das Projekt untersucht die Dynamik photoinduzierter Reaktionen von Einzelmo-lekülen, die in organischen Umgebungen eingebettet sind. Dabei soll insbeson-dere der Einfluß der spezifischen organischen Umgebung auf den Prozeßablauf betrachtet werden. Als Modell einer organischen Umgebung dienen Calixarene; als Benchmark-Beispiel einer photoinduzierten Reaktion wird die Photodissozia-tion von Methyljodid betrachtet. Dabei wird der Einfluß der System-Umgebungs-Wechselwirkung auf die Struktur des Wirt-Gast-Systems und den Ablauf des Dissoziationsprozesses mittels genauer quantenchemischer und numerisch ex-akter quantendynamischer Rechnungen in voller Dimensionalität untersucht. Das Verständnis der Wirt-Gast-Wechselwirkung und deren Auswirkung auf die Reaktionsdynamik sind zentrale Projektziele. • Resultate • Charakterisierung der Wirt-Gast-Wechselwirkung in Calixarenen • Struktur und harmonisches Kraftfeld des Methyljodid-Calixaren-Komplexes auf RI-MP2 -Niveau (mit einer SV(P)-Basis) berechnet. • Wechselwirkungen zwischen Calixaren und verschiedenen Gästen wurden mittels DFT- und RI-MP2-Rechungen untersucht [1,2,6,7] (in Kooperation mit Teilprojekt K5). • Quantendynamik des Photodissoziationsprozesses • Volldimensionale (189D) Quantendynamiksimulationen für das Methyljodid-Calixaren-Gesamtsystem mit der multi-layer MCTDH-Methode sind in Vorbereitung. • Simulation zeitaufgelöster ESCA-Spektren • Entwicklung eines effizienten Koordinatensystems und kin. Energieoperators zur Be-schreibung von Methyl-Gruppen in Quantendynamikrechnungen [4,5]. • Weiterentwicklung der MCTDH/CDVR-Methode zur Durchführung hochdimensionaler Wellenpaketdynamikrechnungen auf allgemeinen gekoppelten elektronischen Potential-flächen [3,5]. • Volldimensionale (9D) Quantendynamikrechnungen für (isoliertes) CH3I abgeschlossen. • Photoionisationsenergien von CH3I als Funktion des C-I-Kernabstandes, (relativistische) ADC(3)-Rechnungen in Kooperation mit M. Pernpointer (Universität Heidelberg):geringe Zeitabhängigkeit der chemischen Verschiebung theoretisch erwartet (s. Experimente in Teilprojekt D4). • Kooperationen im SFB • Teilprojekte K5 und D4 siehe oben, verwandte Fragestel-lungen bzgl. photoinduzierter Prozesse oder numeri-scher Simulationstechniken werden in Teilprojekten D10, K8 bzw. A2, A4, K7, D2 untersucht. Publikationen (Projektmitarbeiter und –leiter blau, Kooperationspartner grün markiert) [1] A. B. Rozhenko, W. W. Schoeller, M. C. Letzel, B. Decker, C. Agena, J. MattayJ. Mol. Str.: THEOCHEM, 732, 7-20 (2005) [2] A. B. Rozhenko, W. W. Schoeller, M. C. Letzel, B. Decker, C. Agena, J. MattayChem. Eur. J., 12, 8995-9000 (2006). [3] A. Viel, W. Eisfeld, S. Neumann, W. Domcke, U. MantheJ. Chem. Phys., 124, 214306 (2006). [4] C. Evenhuis, G. Nyman, U.MantheJ. Chem. Phys., 127, 144302 (2007).[5] A. Viel, W. Eisfeld, C.R. Evenhuis, U. MantheChem. Phys., 347, 331-339 (2008). [6] I. Stoll, R. Brodbeck, S.Wiegmann, J. Eberhard, S. Kerruth, H.-G. Stammler, J. MattayEur. J. Org. Chem., in press (2008). [7] T. Schröder, R. Brodbeck, M. C. Letzel, A. Mix, B. Schnatwinkel, M. Tonigold, D. Volkmer, J. MattayTetra. Lett., in press (2008).
C-I-stretch H3C-I bend Universität Bielefeld SFB 613 K6 Zusatzinformation Anhang Detailierte Resultate zur Quantendynamik des CH3I−Photodissoziationsprozesses hn Vergleich der Gasphasen-Absorptionsspektren für isoliertes Methyljodid: Experiment, ältere (5D) Quantendynamikrechnungen von Hammerich et al. auf einer Vorgänger-PES und aktuelle Resultate. Konische Durchschneidung der adiabatischen Potentialflächen der angeregten elektronischen Zustände 3Q0 und 1Q1 von Methyljodid nahe des Franck-Condon-Gebiets → starke vibronische Kopplung und ultraschnelle nichtadiabatische Dynamik Experimentelle I* / I−Produktverhälnisse: 70% bei 248 nm, 75% bei 266 nm → I-Kanal wird von Rechnungen auf der gegenwärtigen PES unterschätzt Berechnete Schwingungsverteilung der Umbrella-Schwingung im I*-Kanal deutlich zu heiß → vermutlich schlechte Beschreibung des langreichweitigen Bereichs der I*-Fläche durch die genutzte PES Form der von Amatatsu et al. gewählten analytischen Ausdrücke zur 9D-Potentialbeschreibung problematisch → neue, genauere CH3I-Potentialfläche dringend erforderlich Beiträge der I und I* -Kanäle zum Gesamtspektrum






















