Download

1 / 42
420 likes | 473 Views
Structural Bioinformatics. Proteins Structure Predictions. Reminder. 3.1 Final date to chose a project 10.1 Submission project overview (one page) -Title -Main question -Major Tools you are planning to use to answer the questions 11.1 /18.1– meetings on projects

E N D
Structural Bioinformatics ProteinsStructure Predictions
Reminder 3.1 Final date to chose a project 10.1 Submission project overview (one page) -Title -Main question -Major Tools you are planning to use to answer the questions 11.1 /18.1– meetings on projects 9.3 Poster submission 16.3 Poster presentation
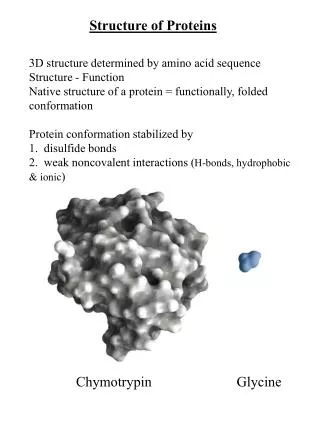
The first high resolution structure of a protein-myoglobin Was solved in 1958 by Max Perutz John Kendrew of Cambridge University. (Won the 1962 and Nobel Prize in Chemistry) In 22.12.2015 there were 114,402 protein structures in the protein structure database.
The 3D structure of a protein is stored in a coordinate file Each atom is represented by a coordinate in 3D (X, Y, Z)
MERFGYTRAANCEAP…. >10,000,000 >100,000 Predicting the three dimensional structure from sequence of a protein is very hard (some times impossible) However we can predict with relative high precision the secondary structure What can we do to bridge the gap??
What do we mean bySecondary Structure ? Secondary structure are the building blocks of the protein structure: =
What do we mean bySecondary Structure ? Secondary structure is usually divided into three categories: Anything else – turn/loop Alpha helix Beta strand (sheet)
The different secondary structures are combined together to form theTertiary Structure of the Proteins
Secondary Tertiary ? ? RBP ? Globin
Secondary Structure Prediction • Given a primary sequence ADSGHYRFASGFTYKKMNCTEAA what secondary structure will it adopt (alpha helix, beta strand or random coil) ?
Secondary Structure Prediction Methods • Statistical methods • Based on amino acid frequencies • HMM (Hidden Markov Model) • Machine learning methods • SVM , Neural networks
Statistical Methods for SS prediction Chou and Fasman (1974) Name P(a) P(b) P(turn) Alanine 142 83 66 Arginine 98 93 95 Aspartic Acid 101 54 146 Asparagine 67 89 156 Cysteine 70 119 119 Glutamic Acid 151 037 74 Glutamine 111 110 98 Glycine 57 75 156 Histidine 100 87 95 Isoleucine 108 160 47 Leucine 121 130 59 Lysine 114 74 101 Methionine 145 105 60 Phenylalanine 113 138 60 Proline 57 55 152 Serine 77 75 143 Threonine 83 119 96 Tryptophan 108 137 96 Tyrosine 69 147 114 Valine 106 170 50 The propensity of an amino acid to be part of a certain secondary structure (e.g. – Proline has a low propensity of being in an alpha helix or beta sheet breaker) Not very useful for predictions
HMM (Hidden Markov Model) An approach for predicting Secondary Structure considering dependency between the position • HMM enables us to calculate the probability of assigning a sequence to a specific secondary structure TGTAGPOLKCHIQWML HHHHHHHLLLLBBBBB p = ?
Beginning with an α-helix The probability of observing Alanine as part of a β-sheet α-helix followed by α-helix The probability of observing a residue which belongs to an α-helix followed by a residue belonging to a turn = 0.15 Table built according to large database of known secondary structures
Example What is the probability that the sequence TGQ will be in a helical structure?? TGQ HHH p = 0.45 x 0.041 x 0.8 x 0.028 x 0.8x 0.0635 = 0.0020995 • What can we learn from secondary structure predictions??
Mad Cow DiseasePrPc to PrPsc PRPc PRPsc
Predicting 3D Structurebased on homology Comparative Modeling/homology modeling Similar sequences suggests similar structure
Sequence and Structure alignments of two Retinol Binding Protein
How do we evaluate structure similarity?? Structure Alignment
Structure Alignments There are many different algorithms for structural Alignment. The outputs of a structural alignment are a superposition of the atomic coordinates and a minimal Root Mean Square Distance (RMSD) between the structures.
The RMSD of two aligned structures indicates their divergence from one another. Atom N (x, y, z) Atom N (x, y, z) Atoms in Protein V Atoms in Protein W Low values of RMSD mean similar structures
Different sequences can result in similar structures 1ecd 2hhd RMSD<1
We can learn about the important features which determine structure and function by comparing the sequences and structures ?
Where are the gaps?? The gaps in the pairwise alignment are mapped to the loop regions
retinol-binding protein odorant-binding protein apolipoprotein D How are remote homologs related in terms of their structure? RBD b-lactoglobulin
PSI-BLAST alignment of RBP and b-lactoglobulin: iteration 3 Score = 159 bits (404), Expect = 1e-38 Identities = 41/170 (24%), Positives = 69/170 (40%), Gaps = 19/170 (11%) Query: 3 WVWALLLLAAWAAAERD--------CRVSSFRVKENFDKARFSGTWYAMAKKDPEGLFLQ 54 V L+ LA A + S V+ENFD ++ G WY + K Sbjct: 1 MVTMLMFLATLAGLFTTAKGQNFHLGKCPSPPVQENFDVKKYLGRWYEIEKIPASFE-KG 59 Query: 55 DNIVAEFSVDETGQMSATAKGRVRLLNNWDVCADMVGTFTDTEDPAKFKMKYWGVASFLQ 114 + I A +S+ E G + K V + ++ +PAK +++++ + Sbjct: 60 NCIQANYSLMENGNIEVLNKELSPDGTMNQVKGE--AKQSNVSEPAKLEVQFFPL----- 112 Query: 115 KGNDDHWIVDTDYDTYAVQYSCRLLNLDGTCADSYSFVFSRDPNGLPPEA 164 +WI+ TDY+ YA+ YSC + ++ R+P LPPE Sbjct: 113 MPPAPYWILATDYENYALVYSCTTFFWL--FHVDFFWILGRNPY-LPPET 159
The Retinol Binding Protein b-lactoglobulin
Taken together MERFGYTRAANCEAP…. FUNCTION
Comparative Modeling Similar sequence suggests similar structure Builds a protein structure model based on its alignment (sequence) to one or more related protein structures in the database
Comparative ModelingGeneral algorithm Modeling of a sequence based on known structures Consist of four major steps : • Finding a known structure(s) related to the sequence to be modeled (template), using sequence comparison methods such as PSI-BLAST 2. Aligning sequence with the templates 3. Building a model 4. Assessing the model
Comparative Modeling • Accuracy of the comparative model is usually related to the sequence identity on which it is based >50% sequence identity = high accuracy 30%-50% sequence identity= 90% can be modeled <30% sequence identity =low accuracy (many errors) However other parameters (such as identify length) can influence the results
What is a good model? ModBase- for homology modelling https://modbase.compbio.ucsf.edu/
Alpha Helix: Pauling (1951) • A consecutive stretch of 5-40 amino acids (average 10). • A right-handed spiral conformation. • 3.6 amino acids per turn. • Stabilized by Hydrogen bonds 3.6 residues 5.6 Å
Beta Strand: Pauling and Corey (1951) β -strand > An extended polypeptide chains is called β –strand (consists of 5-10 amino acids > The chains are connected together by Hydrogen bonds to form b-sheet β -sheet
Loops • Connect the secondary structure elements (alpha helix and beta strands). • Have various length and shapes.