Download

1 / 39
420 likes | 974 Views
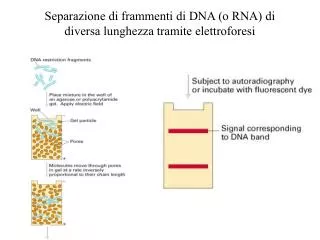
Analisi elettroforetica delle proteine CAPITOLO 10 nuovo testo Capitolo 12 vecchio testo. Tramite ELETTROFORESI le proteine si possono separare in base a una o piu’ delle seguenti caratteristiche : dimensione , carica o idrofobicita’ relativa.

E N D
Analisi elettroforetica delle proteine CAPITOLO 10 nuovo testo Capitolo 12 vecchio testo. Tramite ELETTROFORESI le proteine si possono separare in base a una o piu’ delle seguenti caratteristiche : dimensione , carica o idrofobicita’ relativa.
La separazione elettroforetica si effettua su una matrice solida poiché durante la migrazione in un campo elettrico si generano forze convettive e di diffusione Perché si usa una matrice solida per alcuni tipi di elettroforesi? Da cosa sono generate queste forze?
Le proteine sono cariche ad un pH differente dal loro punto isoelettrico Punto isoelettrico (pI)e’ il valore di pH al quale il numero delle cariche negative sulla molecola prodotta dalla ionizzazione del gruppo carbossilico risulta uguale al numero delle cariche positive acquisite dal gruppo amminico
Le molecole cariche migrano in un campo elettrico in maniera dipendente dalla loro densita’ di carica. Al punto isoelettrico la molecola e’ elettroforeticamente immobile . Una proteina carica che è posta in un campo elettrico ha una distanza di migrazioneproporzionale sia alla intensità di corrente (I) che al tempo (t ).
La correntenella soluzione tra gli elettrodi e’ condotta principalmente dagli ioni del tampone di corsa e solo in piccola parte dagli ioni del campione. La legge di Ohm esprime la relazione tra corrente (I), voltaggio (V), e resistenza (R): R = V / I SI può quindi accellerare una separazione elettroforetica aumentando il voltaggio applicato, MA…….
Durante l’elettroforesi la potenza(W= watts) generata nel mezzo di supporto e’ data da : W = I2 R R= resistenza I= intensità della corrente Aumentando il voltaggio necessariamente si genera CALORE La maggior parte della potenza sviluppata durante elettroforesi viene dissipata in calore
Problemi derivanti dallo sviluppo di calore durante elettroforesi. 1.Maggioretasso di diffusione del campione e degli ioni del buffer =allargamento delle bande da separare 2. Formazione di correnti convettive =possono causare un miscelamento del campione 3.Instabilita’ termica dei campioni=denaturazione delle proteine sensibili al calore conseguente perdita di attivita/folding 4.Diminuita viscosita’ del buffer= riduzione della resistenza del mezzo
Gel di agarosio i pori sono formati da molecole di polisaccaridi che partecipano alla formazione di strutture a doppia elica Questi pori non hanno una struttura regolare la dimensione dei porisi controlla variando la concentrazione di agarosio: piu’ grande e’ il numero delle eliche formate per unita’ di spazio e piu’ piccola sara’ la dimensione media dei pori ,
Gel di poliacrilammide: miscela di bis e monoacrilammide Una singola molecola di bis acrilammide e’ essenzialmente formata da due molecole di acrilammide unite da un gruppo metilico.
La poliacrilammide polimerizzata ha matrice molto regolare con pori di dimensioni uniformi I monomeri di acrilammide formano catene e le molecole di bis-acrilammide danno i metili che formano i ponti cross-lincanti
La polimerizzazione della acrilammide avviene in presenza di TEMED e AMMONIO PERSOLFATO TEMED catalizza la decomposizione dello ione persolfato con la produzione di un radicale libero SO4- .=R* S2O82+ + e- SO42- + SO4- . Il radicale libero (R*= SO4- .) è una molecola con un elettrone spaiato R* sono specie molto reattive. Occasionalmente si procede alla degassazione della acrilammide mix poiche’ l’ ossigeno rimuove i radicali
R*+M RM*+M RMM* Durante le polimerizzazione Il radicale libero (R*= SO4- .), che è una molecola con un elettrone spaiato, reagisce con M (monomero di acrilammide) e forma un legame singolo condividendo il suo elettrone spaiato con uno proveniente dal guscio esterno della molecola del monomero …e così via
PolyAcrylamide Gel Electrophoresis (PAGE) Separazione dellle proteine tra 5 to 2,000 kDal e’ stata introdotta da by Raymond and Weintraub (1959).. La dimensione dei pori si puo’ controllare variando la percentuale di acrilammide e/o Bis-acrilammide (da 3% a 30%),
SDS –PAGE (gel denaturante) Ildetergente anionico SDS = CH3-(CH2)10-CH2OSO3-Na+ Ildetergente anionico SDS distrugge i legami idrogeno, blocca le interazioni idrofobiche e sostanzialmente denatura le molecole proteiche minimizzando così le differenze dovute alla forma molecolare eliminando le strutture secondarie e terziarie. http://www.bio.davidson.edu/courses/genomics/method/SDSPAGE/SDSPAGE.html#SDS Le proteine possono essere completamente denaturate quando sostanze riducenti quali DTT e’ utilizzato insieme al SDS.
La maggioranza delle proteine lega 1.4g SDS per grammo di proteina Il legame del SDS alle proteine causa un’ efficace mascheramento delle cariche intrinseche della catena polipeptidica e apporta una carica netta negativa proporzionale alla lunghezza del polipeptide
SDS-PAGE il miscuglio proteico si separa secondo l’effettivo raggio molecolare (Mr) di ciascuna proteina, che e’ approssimativamente uguale alla dimensione molecolare di ciascun polipeptide. risultato sarà la separazione delle proteine per setacciamento attraverso i pori del gel di poliacrilammide http://www5.amershambiosciences.com/aptrix/upp00919.nsf/Content/Elpho_1D_SDS+PAGE
TAMPONI usati per SDS-PAGE Esistono due tipi di sistemi di buffers: continui e discontinui
run Caricamento Stacking Separating Buffer discontinuo di Ornstein and Davis (1964) modificato da Laemnli Laemmli buffer E’ il piu’ comunebuffer utilizzato per SDS-PAGE gels Laemmli buffere’ costituito da a) Stacking (o gel di impaccamento) con grandi pori b)Separating gel (12%, 10%, 6% etc.). I
Loading RUN Stacking gel ha pori di grandi dimensioni (4% acrilammide) che permettono alle proteine di muoversi liberamente e di concentrarsi sotto l’ effetto del campo elettrico. Lo scopo del gel di impaccamento è di concentrare le proteine in un banda sottile prima che ilcampione entri nel gel separatore
NELLO STACKING GEL Una volta applicata la corrente.. le bande di proteine si assottigliano a causa del fatto che gli ioni di glicinato (carichi -)presenti nel tampone elettroforeticohanno mobilità elettroforetica più bassa dei complessi proteina-SDS ioni di glicinato<complessi proteina-SDS< ioni cloruro (Cl-) I complessi proteina-SDS a loro volta hanno una mobilità elettroforetica, inferiore a quella degli ioni cloruro (Cl-) presenti nel tampone di caricamento (sample buffer)
Il gel di separazione(separating gel) ha pH 8.8 mentre quello di impaccamento (stacking gel) ha pH 6.8. Separazione Separating gel Raggiunto il gel di separazione, il glicinato a causa dell’ ambiente a pH maggiore diventa completamente ionizzato e cresce la sua mobilita’elettroforetica
Il risultato e’ che nel gel separante i complessi proteins_SDS (carichi negativamente) si muovono verso il polo positivo in funzione del setaccio molecolare operato dal gel di poliacrilammide quindi in funzione del loro peso molecolare (MW=kDal) -180 -65 -20 Cioè: le proteine piu’ piccole migreranno piu’ velocemente di quelle piu’ grandi a che saranno rallentate da resistenze frizionali
ALTRI TIPI DI GEL DI POLIACRILAMMIDE a) GEL NATIVO SDS è assente e le proteine non vengono denaturate prima del caricamento Le proteine migrano in dipendenza della loro carica b) GEL a GRADIENTE (tipicamente 5% a 25% ) Permette una maggiore separazione delle proteine con massa simile
con Comassie Blue Il colorante Comassie brilliant blue-G250 (CBB) si lega alle proteine tramite interazioni elettrostatiche dei gruppi sulfonici del colorante. La colorazione CBB visualizza bande con una concentrazione di circa 0.1µg di proteina.
Western blotting trasferimento su membrana di nitrocellulosa (NC) o polyvinylidene fluoride (PVDF) delle proteine separate tramite elettroforesi
APPARATI di traferimento Liquido e SEMI-DRY
Direzione di migrazione 3MM APPARATO DI TRASFERIMENTO Semi-dry
Corrente applicata Nel semi-dry si suggerisce di applicare 1mA/cm2 di membrana in ogni caso mai piu’ di 5mA/cm2 senza refrigerazione! Attenti alle bolle d’aria e all’essicamento della membrana
BUFFER di TRASFERIMENTO : Ruolo del metanolo e del SDS. • Il metanolo presente nel buffer fa : • 1.decrescere la mobilita’ delle proteine durante l’ elettroforesi. • dissociare l’ SDS dalle proteine • aumentare le interazioni idrofobiche tra proteine e membrana. • Denaturare le proteine ad alto peso molecolare e quindi rende il loro passaggio dal gel alla membrana piu’ difficile. Le proteine piccole non sono invece influenzate dalla presenza di metanolo.
L’SDS da una carica negativa alla proteina aiuta il traferimento delle proteine dal gel alla membrana NOTA CHE: L’ SDSagisce in maniera opposta al metanolo per quel che rigurda il suo effetto sulla quantità di proteina che viene traferita dal gel alla membrana
Per migliorare il legame delle proteine con la membrana si puo’ aumentare la concentrazione di metanolo (normalmente tra 10%-20%) oppure ridurre la sua concentrazione se le proteine rimangono nel gel. Un’alta concentrazione di SDS, sebbene faciliti il trasferimento, può interferire con il legame delle proteine alla membrana
APPARATI di traferimento Liquido Piu’ tempo le proteine sono a contatto con la membrana e piu’ facilmente vi si legheranno Apparato liquido +buffer +tempo di corsa rispetto ad un semi-dry Quando si usa? A) Si devono trasferire proteine ad alto peso molecolare poichè necessitano di piu’ tempo per essere trasferite e di maggiori quantita’ di buffer B) Le proteine sono corse su gel ad alta concentrazione di acrilammide o molto spessi.
Tempi di trasferimento I tempi di trasferimento variano in dipendenza dalla dimensione delle proteine da trasferire, tipo di membrana e tipo di apparato per blotting utilizzato semi-dry : max 2h o liquido : fino a 28h
TIPI di MEMBRANE per il trasferimento di proteine: Le membrane di PVDF lega le proteine principalmenteattraverso interazioni idrofobiche. sono comunemente usate per la loro i resistenza chimica e per la stabilita’ fisiologica. La nitrocellulosa (NC) lega le proteine primariamente tramiteinterazioni elettrostatiche o idrofiliche
Membrane PVDF : legano le proteine principalmenteattraverso interazioni idrofobiche. Esistono diversi tipi di membrane PVDF Diversa porosità e resistenza Membrane derivate con procedure che aggiungono cariche alla membrana PVDF in modo da permettere oltre che interazioni idrofobiche anche interazioni elettrostatiche. Membrane PVDF derivate sono per esempio usate per legare DNA o RNA o per procedure di purificazione a scambio ionico delle proteine .
SVANTAGGI La maggiore limitazione della nitrocellulasa e’ la sua scarsa capacita’ di legare proteine con basso peso molecolare e la fragilità MEMBRANE DI NITROCELLULOSA (NC) Sono meno sensibili della PVDF alle concentrazioni di SDS (eg. danno meno “background signal” ) perchè legano le proteine primariamente tramite interazioni elettrostatiche o idrofiliche. L’ SDS, coprendo le cariche delle proteine, puo’ impedire la formazioni di legami idrofobici con la PVDF ma non interferisce con la formazione deli legami elettrostatici con la NC. Proteine che sono scarsamente trasferibili possono essere meglio trasferite poiche’ si puo’ utilizzare SDS nel buffer di traferimento (normalmente questo non e’ presente o e’ controproducente) .
PRO e contro la PVDF rispetto alla NC. • La PVDF e’ meglio per: • il trasferimento di proteine a basso peso molecolare perchè lega piu’ fortemente le proteine ma • 1) e’ piu’ sensibile alla presenza di impurita’ presenti nel buffer (compreso SDS, glicina e Tris) • la presenza di queste sostanze sulla membrana puo’ dare origine ad un alto “background”. • 2) e’ piu’ difficile eluire le proteine da PVDF • quindi meglio NON usare PVDF in procedure che implicano la eluizione della banda proteica dalla membrana