Download

1 / 68
710 likes | 940 Views
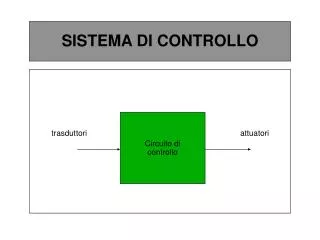
Studi di associazione caso-controllo Dr.ssa Francesca Gensini. Frequenza di tipi diversi di malattie genetiche. Fonte: Rimoin, Connor, Pyeritz (1997) Emery and Rimoin’s Principles of Medical Genetics , 3 ed. Churchill Livingstone, Edinburgh. MALATTIE MULTIFATTORIALI

E N D
Frequenza di tipi diversi di malattie genetiche Fonte: Rimoin, Connor, Pyeritz (1997) Emery and Rimoin’s Principles of Medical Genetics, 3 ed. Churchill Livingstone, Edinburgh
MALATTIE MULTIFATTORIALI Originano dall’interazione tra piùfattori genetici predisponenti e fattori ambientali • - molto più frequenti delle malattie mendeliane monogeniche • - rappresentano una delle maggiori cause di morbilità cronica e di mortalità nella popolazione generale • importanza dello studio dei fattori genetici predisponenti per: • migliore conoscenza dei meccanismi patogenetici coinvolti • migliori approcci terapeutici
Fattori complicanti l’analisi delle componenti genetiche dei caratteri multifattoriali Diversi loci implicati, perlopiù con effetto “debole” Eterogeneità genetica: esistenza di differenti mutazioni (anche in geni diversi) che causano fenotipi simili Scarsa conoscenza ed eterogeneità di esposizione ai fattori ambientali
Studi di associazione (caso/controllo) generalmente impiegati per testare l’ipotesi del coinvolgimento di geni candidati* nella suscettibilità ad una malattia *geni che codificano per proteine di cui si conosca o si sospetti un coinvolgimento nel processo patologico STRATEGIE PER L’IDENTIFICAZIONE DEI GENI IMPLICATI NELLA GENESI DEI CARATTERI MULTIFATTORIALI Analisi di linkage (famiglie) generalmente impiegati per la ricerca di nuovi geni
Problemi degli studi di linkage nelle mal. multifattoriali 1- difficoltà di trovare famiglie numerose e con vari casi per l’insorgenza tardiva delle malattie e alta mortalità 2- poco efficaci nell’identificazione di varianti genetiche con effetto debole 3- rispetto alle malattie monogeniche deve essere studiato un maggior numero di famiglie con un alto numero di marcatori del DNA
Dagli studi di linkage all’identificazione dei geni di suscettibiltà… • Esame più in dettaglio delle regioni cromosomiche identificate: • sono molto ampie e contengono centinaia di geni: ciascuno di essi potrebbe essere il responsabile • Aumentare il numero di famiglie studiate Analisi combinata dei dati di diversi gruppi di ricerca • Studio di individui affetti con anomalie cromosomiche che coinvolgono queste regioni • Studio di geni candidati • Studi di associazione allelica / Linkage disequilibrium mapping
studio di 2 popolazioni distinte di soggetti: 1- CASI:presenza della malattia 2- CONTROLLI: assenza della malattia STUDI DI ASSOCIAZIONE (CASO/CONTROLLO)(i più utilizzati nello studio delle mal.multifattoriali) Si utilizzano singoli soggetti (non sono necessari nuclei familiari)
Identità genetica umana • 99.9% di identità genomica • 0.01% di differenze interindividuali (polimorfismi)
LOCUS: regione cromosomica unica che corrisponde ad un gene o a qualche altra sequenza di DNA • ALLELE: una o più forme alternative di un gene o di una sequenza di DNA
Eterozigote: individuo con alleli diversi allo stesso locus (CT) • Omozigote: individuo con alleli identici ad uno stesso locus (CC o TT) • Genotipo: costituzione genetica di un individuo o più specificamente gli alleli presenti ad ogni particolare locus • Mutazione: alterazione della sequenza nucleotidica di una molecola di DNA • Polimorfismo: variazione della sequenza nucleotidica di un gene, senza effetto fenotipico, che esiste nella popolazione con una frequenza di almeno l’1%
Single Nucleotide Polymorphism (SNP) • Il più comune tipo di polimorfismo • Più di 10 milioni di SNPs nell’uomo GATTTAGATCGCGATAGAG GATTTAGATCTCGATAGAG
SNPs Nelle regioni codificanti: -causa di malattia mendeliana se alterano la funzione o la struttura della proteina -importanti in farmacogenomica se alterano la struttura di una proteina coinvolta nel metabolismo di un farmaco -causa di mutazioni missenso in regioni codificanti possono aumentare il rischio di ammalarsi per patologie comuni Nelle regioni con elementi regolatori: -alterando la trascrizione possono essere associati ad un aumento del rischio per patologie comuni Nelle regioni non codificanti: -non hanno impatto diretto sul fenotipo dell’individuo, utili negli studi di popolazione
Control Disease Allele 1 Allele 2 Marker A: Allele 1 = Allele 2 = Marker A is associated with Phenotype STUDI DI ASSOCIAZIONE: confronto della frequenza di un polimorfismo tra un gruppo di casi e un gruppo di controlli
Distribuzione allelica Esempio: Polimorfismo A/G (genotipi AA-AG-GG) controlli pazienti 70 78 A 74 G 106 Se un particolare allele mostra una frequenza maggiore nei casi rispetto ai controlli vi è evidenza di associazione Test chiquadrato=3.935 p=0.04 O.R. 1.5961
CALCOLO DELL’ODDS RATIO (O.R.) “a”= numero dei pazienti con l’allele X, “b”= numero dei controlli con l’allele X, “c”= numero dei pazienti senza l’allele X, “d”= numero dei controlli senza l’allele X. Si calcola quanto maggiore è la probabilità di avere l’allele X piuttosto che non averlo per i pazienti (a/a+c):(c/a+c) = a/c, rispetto a quanto minore sia la probabilità di avere l’allele X piuttosto che non averlo per i controlli (b/b+d):(d/b+d) = b/d. OR= a/c : b/d = ad/bc Il valore ottenuto indica la forza dell’associazione e può assumere valori da 0 a +∞. OR=1 indica che non c’è associazione.
paz controlli A/A 8 20 A/G 54 38 G/G 26 18 X2=8,548 2df, p=0.0139 Distribuzione dei genotipi
Studi che confrontano la frequenza di un polimorfismo tra un gruppo di interesse (pazienti; soggetti con determinati tratti fenotipici) e un gruppo di controllo Quando una variante allelica è più frequente nei casi rispetto ai controlli, si dice che è “associata” con il fenotipo.
Cause di associazione allelica Effetto diretto della variante genetica sul fenotipo 2. La variante può essere vicina (in linkage disequilibrium*) ad un’altra variante responsabile del fenotipo. 3.Associazioni “spurie” dovute a diversi eventi: per es. presenza di stratificazione nella popolazione marcatore Allele di suscettibilità
Aplotipo: particolare combinazione di varianti allelichein una specifica regione di un cromosoma
SNP2 SNP1 SNP1 SNP2 Soggetto 1 A G Soggetto 1 A G Soggetto 2 A G Soggetto 2 A C Soggetto 3 A G Soggetto 3 A C Soggetto 4 A G Soggetto 4 A G Soggetto 5 T C Soggetto 5 T C Soggetto 6 T C Soggetto 6 T G Soggetto 7 T C Soggetto 7 T G Soggetto 8 T C Soggetto 8 T C LINKAGE EQUILIBRIUM LINKAGE DISEQUILIBRIUM
STUDI DI ASSOCIAZIONE: LIMITI 1- selezione dei pazienti 2-selezione dei controlli ( transmission disequilibrium test) 3- risultati discordanti nelle diverse popolazioni per diverso background genetico 4- necessità di un campione molto numeroso 5- link biologico tra gene candidato e patologia 6- possibilità di testare solo un polimorfismo già conosciuto 7- assenza di associazione non esclude il gene candidato 8- necessità di analizzare > 1 polimorfismo 9- necessità di analisi statistica adeguata
Studi di associazione con controlli interni: TDT: test di disequilibrio della trasmissione (transmission disequilibrium test) si analizza il probando + 2 genitori (problema per le malattie ad esordio tardivo) si propone di identificare la trasmissione preferenziale di un dato allele (X) nei figli affetti di famiglie nucleari (padre-madre-figlio) si selezionano i genitori eterozigoti per l’allele X se un allele predispone alla malattia o è in linkage-disequilibrium con l’allele-malattia, si osserverà una trasmissione preferenziale di tale allele nei figli affetti quale popolazione di controllo viene utilizzata la “popolazione” dei cromosomi non trasmessi ai figli affetti dai propri genitori occorre analizzare il 50% di soggetti in più rispetto ai normali test di associazione
Mendel’s law : Each parental allele carried by an autosome has an equal probability (i.e. 0.5) to be transmitted to an offspring a/b c/d a/c
Transmission of an autosomic multiallelic polymorphism in trios a/b c/d a/d b/d b/b a/d c/b c/a a/b c/d a/c a/d b/a c/c b/c a/c c/d b/c c/d a/d a/d a/b a/d a/b a/d c/d c/d a/c c/d a/a Frequency of transmission of allele a : Fa = 6/12= 0.5
a/b c/d a/d b/d b/b a/d c/b c/a a/a c/d a/c a/d b/a c/a a/c a/c a/c a/c c/d a/d a/d c/b a/d a/b a/d a/c c/d a/a c/a a/a Transmission disequilibrium in affected offsprings Phenocopy Allele a is transmitted with a frequency : Fa = 10/12= 0.83 (p < 0.038)
Studi di associazione con controlli interni: Transmission disequilibrium test The A allele is transmitted to affected offspring four times out of five.
attivazione piastrinica modificazioni del flusso ematico disfunzione endoteliale PATOLOGIA VASCOLARE attivazione della coagulazione alterazione della fibrinolisi Fattori di rischio classici: - ipertensione, diabete,fumo, iperlipidemia - intervento chirurgico, immobilizzazione, gravidanza, ter. estroprogestinica Fattori di rischio emergenti: ……… fattori genetici Scelta di geni candidati
SISTEMA RENINA ANGIOTENSINA E NITROSSIDO Angiotensinogeno Renina Bradichinina Angiotensina I ACE Nitrossido sintasi Angiotensina II peptidi inattivi Nitrossido - vasocostrizione - proliferazione c.m.l.vasali - adesione e aggregazione piastrinica - espressione Fattore Tissutale - produzione t-PA - produzione di PAI-1 - vasodilatazione - proliferazione c.m.l.vasali - adesione e aggregazione piastrinica - espressione Fattore Tissutale - adesione leucociti all’endotelio coinvolgimento di: endotelio, coagulazione, piastrine, fibrinolisi, flusso ematico
GeneeNOS: polimorfismoG894T (ex. 7) A T G A C C C C G 3’ 5’ A T T G A C C C C 3’ 5’ Mbo I G894T attività enzimatica produzione basale di nitrossido (Veldman BA et al., J Hypertension 2002) GeneeNOS: polimorfismoT-786 C (promotore) T G G C G G C T T 3’ 5’ T G G C G G C T 3’ C 5’ Nae I T-786C attività del promotore (Nakayama M et al., Circulation 1999) GeneeNOS: polimorfismo4a/4b (int. 7) b: variante comune (5 tandem repeats di 27 bp) a: variante rara (4 tandem repeats di 27 bp) 4a/4b livelli plasmatici di nitrossido (Wang XL. et al., ATVB 1997)
METODI Estrazione di DNA genomico da sangue periferico Amplificazione mediante PCR Digestione con enzimi di restrizione ANALISI STATISTICA Conta diretta Test chi-quadrato Analisi di regressione multipla T-test, analisi della varianza p<0,05 considerato statisticamente significativo
478 pazienti con cardiopatia ischemica M=69.5%, F=30.5%; età 68 (26-87) Infarto miocardico n=224 (47%), Angina n=254 (53%) Mal. monovasale n=217 (45%), Mal. plurivasale n=261 (55%) 538 controlli M=68.8%, F=31.2%; età 66 (33-89) Fattori di rischio classici Pazienti Controlli Ipertensione 57.3% 30.1% Dislipidemia 52.5% 19.9% Diabete 29.5% 3.9% Fumo 48.9% 21.6% Familiarità 33.2% 17.5%
polimorfismo T-786C gene eNOS 80% Pazienti Controlli p<0.0001 p<0.0001 60% 40% 20% TT allele C CC TC
polimorfismo G894T gene eNOS Pazienti Controlli 80% p=0.01 p=0.02 60% 40% 20% GG allele T TT GT
polimorfismo 4a/4b gene eNOS Pazienti Controlli 80% p=0.02 p=0.005 60% 40% 20% 4b4b allele4a 4a4a 4a4b
1.7 (1.2-2.3) p<0.001 Analisi univariata -786CC vs TC+TT 1.6 (1.1-2.3) p=0.008 894TT vs GT+GG 1.9 (1.01-3.9) p=0.04 Analisi Multivariata* *Corretta per ipertensione, dislipidemia, fumo, diabete 4a4a vs 4a4b+4b4b 1.4 (0.9-2.1) p=ns -786CC vs TC+TT 1.5 (0.9-2.5) p=ns 894TT vs GT+GG 2.5 (1.1-5.4) p=0.02 4a4a vs 4a4b+4b4b 6.1 (1.3-29.6) p=0.02 4a4a / -786CC Infarto 3.6 (1.2-11.5) p=0.03 4a4a vs 4a4b+4b4b 1.4 (0.6-3.2) p=0.5 Angina 4a4a vs 4a4b+4b4b 0 1 2 3 4 5 20 Odds ratio (IC95%)
Whole Genome Associations Disease Population N=500 Matched Control Population N=500 • ~3,000,000 common SNPs across genome • Representing every gene 22 1 Regions of association P value 1 22 Chromosomal Location Informatics to identify gene(s) mapped to associated SNP
Caratterizzazione di nuovi SNPs + sviluppo di nuove tecnologie Obiettivo: ridurre il numero totale degli SNP da analizzare mantenendo un potere analitico sufficiente 2 approcci: 1- analisi limitata ai SNP localizzati all’interno di sequenze codificanti 2- esplorare indistintamente regioni geniche e intergeniche
1- analisi limitata ai SNP localizzati all’interno di sequenze codificanti Basato sull’assunto che le varianti genetiche che influenzano la suscettibilità a malattie genetiche dovrebbero determinare effetti funzionali sulla proteina Numero di SNPs da analizzare ~50000-100000 MA: . Selezione degli SNPs sulla base degli effetti funzionali non ancora completata . Molte varianti hanno bassa frequenza e quindi sarebbe necessario aumentare la dimensione del campione da studiare
2- esplorare indistintamente regioni geniche e intergeniche Circa il 65-85% del genoma umano è organizzato in blocchi inscindibili di oltre 10000bp. Ciascuno di questi blocchi aplotipici può contenere 12 o più SNPs che tenderanno ad essere trasmessi insieme nelle generazioni. Si cerca di determinare per ciascun blocco, gli SNPs (tagSNPs) che lo identificano Sfrutta l’esistenza dei blocchi di linkage disequilibrium L’analisi è ridotta ai soli haplotype tagging SNPs (~300000-600000)
L’associazione di uno di questi blocchi, identificato per mezzo dei suoi tagSNPs, e una data patologia, rivelerebbe in quale regione del genoma ricercare il gene responsabile della suscettibilià alla malattia in esame MA: . maggior numero di SNP da analizzare . è fondamentale che i blocchi aplotipici siano definiti nelle popolazioni cui appartengono i campioni in esame
MALATTIE INFIAMMATORIE INTESTINALI • Morbo di Crohn • Colite Ulcerosa • Forme miste
MALATTIE INFIAMMATORIE INTESTINALI -Concordanza in gemelli (dati del Registro Svedese): CD: 8/18 MZ e 1/26 DZ concordanti CU: 1/16 MZ e 0/20 DZ concordanti -Familiarità: s 15-40 per CD (rischio relativo generico per IBD, in particolare per CD), 8-10 per CU -Prevalenza elevata in particolari gruppi etnici: negli Ebrei Ashkenaziti CD 2-8X rispetto alla popolazione generale (40-250/100.000 per CD; 100-300/100.000 per IBD nel complesso) Evidenze in favore del coinvolgimento di fattori genetici
Genetica del Morbo di Crohn (CD)Hugot et al. Nature 379:821-823, 1996 • Analisi di linkage “genome-wide” • Due pannelli di famiglie con CD puro: • pann.1: 25 fam. • pann.2: 53 fam. • Dimostrata evidenza di linkage conmarcatori della regione pericentromerica del cromosoma 16 in entrambi i pannelli (nel primo pannello anche linkage con un marcatore del cromosoma 1, non confermato nel secondo)
Analisi della condivisione di alleli tra coppie di fratelli CD (ASP) Fraz. media alleli condivisi 0.55 0.58 0.58 0.58 0.57 0.55 0.58 0.51 Locus D16S24 SPN D16S409 D16S419 D16S408 D16S514 D16S503 D16S421 T-test 1.72 2.36 3.41 3.05 3.09 1.69 2.44 0.3 p 0.044 0.0099 0.0004 0.0014 0.0012 0.0471 0.0082 >0.05 Hugot et al. Nature 379:821-823, 1996
Morbo di Crohn e cromosoma 16 Fraz. media alleli condivisi 0.58 0.58 - - - 0.72 0.56 - - - 0.58 LOD Score Multipoint 3.17 2.41 2.6 2.02 2.49 6.3 1.71 .25 2.71 .69 5 Locus D16S409-419 D16S411 D16S411 D16S419 D16S769 D16S409 D16S409-411 D16S753 D16S408 D16S411 D16S411 Autori Hugot et al. 1996 Ohmen et al. 1996 Parkes et al. 1996 Mirza et al. 1998 Brant et al. 1998 Cavanaugh et al. 1998 Curran et al. 1998 Rioux et al. 1998 Annese et al. 1999 Vermeire et al. 2000 IBD Int. Genet. Cons. 2001 p 0.0004 0.019 0.002 0.02 0.007 0.0000017 0.01 - - .5 -
Morbo di Crohn e Gene NOD2/CARD15(Hugot et al. Nature 411: 599-603Ogura et al. Nature 411:603-606) • Mappatura fine della regione IBD1 sul cromosoma 16 in 235 famiglie (Hugot) • Evidenza di linkage disequilibrium tra nuovi marcatori della regione e CD • Identificazione di polimorfismi associati a CD mediante Pedigree-Disequilibrium Test (PDT) • I polimorfismi associati sono localizzati all’interno del gene NOD2/CARD15 • Analisi diretta del gene candidato NOD2/CARD15 (Ogura)