Download
1 / 64
650 likes | 728 Views
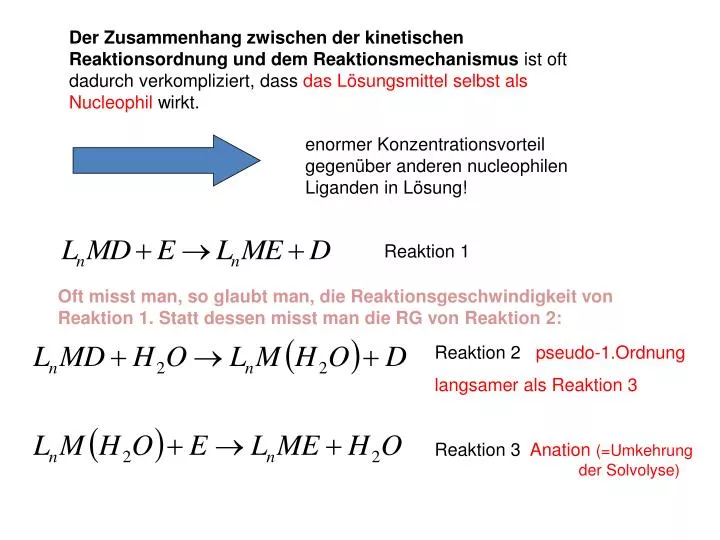
Der Zusammenhang zwischen der kinetischen Reaktionsordnung und dem Reaktionsmechanismus ist oft dadurch verkompliziert, dass das Lösungsmittel selbst als Nucleophil wirkt. enormer Konzentrationsvorteil gegenüber anderen nucleophilen Liganden in Lösung!. Reaktion 1.

E N D
Der Zusammenhang zwischen der kinetischen Reaktionsordnung und dem Reaktionsmechanismus ist oft dadurch verkompliziert, dass das Lösungsmittel selbst als Nucleophil wirkt. enormer Konzentrationsvorteil gegenüber anderen nucleophilen Liganden in Lösung! Reaktion 1 Oft misst man, so glaubt man, die Reaktionsgeschwindigkeit von Reaktion 1. Statt dessen misst man die RG von Reaktion 2: Reaktion 2 pseudo-1.Ordnung langsamer als Reaktion 3 Reaktion 3 Anation (=Umkehrung der Solvolyse)
Ligandensubstitution in oktaedrischen Komplexen • Kinetische Studien an oktaedrischen Komplexen in saurer wässriger Lösung zeigen in den meisten Fällen ein Geschwindigkeitsgesetz 1. Ordnung und keinen Einfluss des eintretenden Liganden auf die Reaktionsgeschwindigkeit • Weist dies auf einen dissoziativen Mechanismus hin?
Ligandensubstitution in oktaedrischen Komplexen: 7 Beweise für den dissoziativen Mechanismus: Elektronische Gründe Einfluss der eintretenden Gruppe auf die Reaktionsgeschwindigkeit (gering!) Einfluss der austretenden Gruppe auf die Reaktionsgeschwindigkeit (groß!) Zusammenhang zwischen Gleichgewichtskonstante und Geschwindigkeitskonstante Einfluss der Ladung des Komplexes Größe der Liganden Aktivierungsparameter
Sterische Gründe • „SterischeAbsättigung“ bei Koordinationszahl Z=6 • Z>6 benötigt eine erhöhte Aktivierungsenergie, dies führt zu einer nicht konkurrenzfähigen langsamen Reaktionsgeschwindigkeit im Falle eines Reaktionswegs über einen siebenfach koordinierten ÜZ • Für Co3+ Komplexe gilt speziell, dass wenn 6 Liganden 6 Elektronenpaare beisteuern, gerade 18 Valenzelektronen vorhanden sind • Ein weiterer Ligand in einem ÜZ mit erhöhter Koordinationszahl würde eine sehr ungünstige elektronische Energie für sein Elektronenpaar vorfinden, siehe oktaedrische Molekülorbitale
aus: William W. Porterfield, Inorganic Chemistry, San Diego, 1993
Geringer Einfluss der eintretenden Gruppe E auf die Reaktionsgeschwindigkeitskonstante • Zu erwarten, wenn die Dissoziation der geschwindigkeitsbestimmende Schritt des Mechanismus ist. • Beispiel: Die Geschwindigkeitskonstanten der Reaktionen von [Co(NH3)5(H2O)]3+ mit Cl-, Br-, NCS-, N3-, NO3-, H2PO4-, NH3 liegen alle sehr nahe beisammen mit k=1.3x10-6 bis k=2.5x10-6 L mol-1 s-1 (25°C) . • Wäre der Prozess assoziativ, dann müssten die Bindungseigenschaften des hereinkommenden Liganden sehr stark die Geschwindigkeitskonstante beeinflussen!
Großer Einfluss der austretenden Gruppe D auf die Geschwindigkeitskonstante • Dann zu erwarten, wenn der Bruch der M-D Bindung der geschwindigkeitsbestimmende Schritt ist. • Kinetische Daten für die saure Hydrolyse von [Co(NH3)5X]2+ zeigen, dass die Geschwindigkeitskonstanten für verschiedene Liganden X über 8 Größenordnungen variieren!
Hydrogenoxalat Trifluormethansulfonat (Triflat) aus: William W. Porterfield, Inorganic Chemistry, San Diego, 1993
Zusammenhang zwischen Gleichgewichtskonstante und Geschwindigkeitskonstante • saure Hydrolyse von [Co(NH3)5X]2+ • ln k1 aufgetragen gegen ln K für verschiedene Liganden X ergibt eine Gerade, mit dem Anstieg 1.03 • d.h. und haben fast den gleichen Wert!
aus: William W. Porterfield, Inorganic Chemistry, San Diego, 1993
aus: William W. Porterfield, Inorganic Chemistry, San Diego, 1993
ÜZ ähnlich dem Reaktionsprodukt! • D.h. beide haben ähnliche Energie und Struktur • Der ÜZ muss also eine Konfiguration des Systems sein, bei der sich das X- Ion bereits vom Co3+ abgetrennt hat • Dissoziativer Mechanismus • Der Unterschied zwischen ÜZ und Produkt ist nur, dass das H2O Molekül noch in der äußeren Koordinationssphäre liegt
Elektrische Ladung des Komplexes • Wenn ein Anion von einem positiv geladenen Komplex abdissoziiert, und die Dissoziation der geschwindigkeitsbestimmende Schritt ist (dissoziativer Mechanismus) so sinkt die Reaktionsgeschwindigkeit mit steigender positiver Ladung des Komplexes. (Wäre der Mechanismus assoziativ, so würde der Komplex mit höherer positiver Ladung den eintretenden Liganden z.B. das polare H2O Molekül oder einen negativ geladenen Liganden, verstärkt anziehen, und die Reaktion würde mit steigender positiver Ladung des Komplexes schneller werden .)
aus: William W. Porterfield, Inorganic Chemistry, San Diego, 1993
Größe des Liganden • Beispiel: methyl-substituierte Ethylendiamin Liganden • Da Methylgruppen nur einen sehr geringen induktiven Effekt (+ I- Effekt) haben, ist der elektronische Unterschied zwischen diesen Liganden gering • Ihre Größe steigt jedoch stark mit steigender Anzahl der Methylgruppen • Größeres Ligandenvolumenmacht es leichter, eine abdissoziierende Gruppe hinauszudrängen • Steigende Reaktionsgeschwindigkeit mit steigendem Liganden-Volumen weist auf einen dissoziativen Mechanismus hin • Wäre der Mechanismus assoziativ, so würde steigendes Ligandenvolumen die Reaktion verlangsamen
aus: William W. Porterfield, Inorganic Chemistry, San Diego, 1993
Aktivierungsparameter und • Sind bei Ligandensubstitution an oktaedrischen Komplexen meist positiv • Dies weist auf einen dissoziativen Mechanismus hin! Co3+ und die meisten anderen okteadrischen Komplexe tauschen ihre Liganden mit einem dissoziativen Mechanismus aus Ausnahmen: Die Wasser-Austauschreaktionen von Cr(H2O)63+ und von Fe(H2O)63+ zeigen ein negatives (Ia Mechanismus)
Weitere Ausnahme: Großes Zentralmetall Die RG hängt davon ab, ob X Chlorid oder Bromid ist (mit Bromid doppelt so schnell) assoziativer Mechanismus
Mechanismus der basischen Hydrolyse • Unterscheidet sich stark von dem der sauren Hydrolyse • Co3+ Komplexe mit NH3 oder Aminen als Liganden hydrolysieren oberhalb pH=4 wesentlich rascher als in sauren Lösungen • Das Geschwindigkeitsgesetz ist immer 2. Ordnung: • Würde auf einen assoziativen Mechanismus hinweisen, dies ist aber nicht der Fall! Beweis für einen dissoziativen Mechanismus: Sterischer Effekt - Je größer der Amin-Ligand, desto rascher die Reaktion.
Wieso kann ein dissoziativer Mechanismus zu einem Geschwindigkeitsgesetz 2. Ordnung führen? • Der NH3 Ligand wird dadurch, dass er an Co3+ koordiniert ist, fähiger, ein H+ abzuspalten • In wässr. Lsg ist das OH- -Ion nicht imstande, NH3 zum NH2- Ion zu deprotonieren. Ist aber das NH3 Molekül an das Co3+ koordiniert, so zieht das positiv geladene Metallion Elektronendichte vom NH3 ab, sodass es saurer wird und ein Amid-Ion entstehen kann • Dies ist sehr oft die Art Änderung der Liganden-Reaktivität, die in Übergangsmetall-Katalysatoren und Metalloenzymen wirksam ist.
Liganden-Substitution unter Beteiligung mehrzähniger Liganden • Der Ersatz von 2 koordinierten Wassermolekülen durch einen zweizähnigen Liganden L-L erfolgt in zwei aufeinanderfolgenden Schritten. • Man kann die Steady-State Näherung auf die Zwischenverbindung anwenden:
a) k2>>k-1 In dem Fall ist kf=k1 • Dann ist die Gesamt-Reaktionsgeschwindigkeit der Chelatbildung durch die Bildung der M-L-L Zwischenverbindung bestimmt und die Reaktion ist durch dieselben Faktoren kontrolliert wie eine Substitutionsreaktion mit einem einzähnigen Liganden. • k bei 25°C für die Komplexbildung von Ni2+ mit py, bpy und tpy sind sehr ähnlich, obwohl Komplexe mit keinem, einem, bzw. zwei Chelatringen gebildet werden. • k=4x103, 1.5x103, 1.4x103 M-1s-1
b) k2<<k-1 In dem Fall ist kf=k1k2/k-1 der geschwindigkeitsbestimmende Schritt ist die Bildung des Chelatringes z.B. wenn die Bildung des Chelatringessterisch gehindert ist Beispiel: bei der Komplexierung von Ni2+ (pH=8; [Ni2+]=100 mM) durch sulfoniertes 2-pyridylazo-1-naphtol ist der Ringschluss sehr langsam (k≈25 s-1) 3 zähniger Ligand, Ringbildung erfolgt durch die ortho-OH Gruppe, die Azo-Gruppe, das heterozyklische N-Atom und das Metallion
Wenn der ringschließende Arm protoniert ist, und interne Wasserstoffbrücken gebrochen werden müssen: Reaktion von protoniertem 3,5Dinitrosalicylat mit Ni2+ k=3.8x102 M-1s-1. • Mit unprotoniertem 3,5 Dinitrosalicylat ist die Reaktion wesentlich rascher, k=3.1x104 M-1s-1 • Anwendung der Steady State Näherung auf die beiden Zwischenverbindungen ergibt: • Die Frage ist, ob der Bruch der H-Brückenbindung in Reaktionsschritt 2 oder der Ringschluss in Reaktionsschritt 3 geschwindigkeitsbestimmend ist. • (Quelle: R.G. Wilkins)
Effekt des pH auf die Geschwindigkeit der Substitution bei Chelatkomplexen • Das Verhalten eines Chelatliganden L und seiner protonierten Form LH+ ist unterschiedlich • Sowohl die Bildung als auch die Hydrolyse von Chelatkomplexen ist daher in der Regel pH-abhängig weil gewöhnlich K1<<1, gilt: K2>>K3 kinetischen Messungen an Metallkomplexen (Ni2+, Cr3+) von Polyaminen zeigten, dass auch k2>>k3 In der umgekehrten Richtung kann ein Proton die Ringöffnung direkt unterstützen, oder auf dem Weg reaktiver protonierter Spezies
Metallionen-unterstützte Dissoziation (Hydrolyse) von Chelatkomplexen • Metallionen können die Dissoziation von Komplexen mit multidentaten Liganden beschleunigen • Der Reaktionsweg ist kompliziert und umfasst zweikernige Zwischenverbindungen, die M und M1 enthalten
D. W. Margerum, D. L. Janes, and H. M. Rosen, Transfer of EDTA from Ni(II) to Cu(II), Journal of the American Chemical Society, 1965
Ligandensubstitution unter Beteiligung von Porphyrinen • Konzept eines Reaktionswegs über einen sitting-atop (SAT) Komplex, wobei das reagierendeMetallionmit den N Atomen des Porphyrinringeswechselwirkt, ohnedass die N-H Gruppendeprotoniertwerden. Die Bildung des SAT KomplexesistdergeschwindigkeitsbestimmendeSchritt. • In einemraschenweiterenSchrittzerfälltder SAT Komplexzu den Produkten, wobei 2 Protonenfreigesetztwerden. L4 M
Der SAT-Komplex wird als mit dem freien Metallion und Porphyrin in einem thermodynamischen Gleichgewicht existierendes Assoziat betrachtet, dessen Bildungskonstante stark mediumabhängig ist. • Bestimmte Gruppen an der Peripherie des Porphyrins erleichtern die Inkorporation des Metallions in den Ring, indem sie mit dem Metallion ein Addukt bilden, das dann weiterreagiert. • Beispiel: tmppH2 reagiert schnell mit Co(II), Cu(II), und Ni(II) in Dimethylformamid)/H2O:
13 aus: Ralph G. Wilkins, MechanismsofReactionsof Transition MetalComplexes, VCH, 1991
Metall-Ionen unterstützte Bildung eines Metalloporphyrins: Route (B) Da man kein freies Porphyrin während des Austausches von M und M* beobachtet, muss es sich um einen assoziativen Prozess handeln. aus: Ralph G. Wilkins, MechanismsofReactionsof Transition MetalComplexes, VCH, 1991
Die Reaktion von Mn2+ mit tppsH24- in Wasser ist sehr langsam. • Die Aufnahme von Mn2+ in den Porphyrinring wird jedoch stark beschleunigt durch die Gegenwart kleiner Mengen von Cd2+ Ionen, die als Katalysator wirken:
Die Reaktion von Cd2+ mit tppsH24- ist rasch, denn das größere Metallion bildet einen out-of-plane Komplex. Dieser kann wiederum leicht durch das stärker bindende Mn2+ attackiert werden. ----------------------------------------------------------------- • Die Dissoziation der meisten Metall-Porphyrine M(II)P ist sehr langsam. • H+ beschleunigen die Entfernung des Metallions. Häufig findet man folgendes Geschwindigkeitsgesetz:
Isobiestische Punkte bei UV-VIS Spektren Isobiestische Punkte sind Wellenlängen, bei denen die Absorption konstant bleibt, während sich die Konzentrationen der Reaktanden und Produkte ändern: weisen darauf hin, dass keine nachweisbaren Zwischenverbindungen auftreten. z.B. Reaktion von Hg(tpp) mit Zn2+ in Pyridin. Hier tritt die freie tpp Base nicht auf, sie hätte ein von beiden Komplexen unterschiedliches Spektrum. aus: Ralph G. Wilkins, MechanismsofReactionsof Transition MetalComplexes, VCH, 1991
Templat-Chemie • Man kann Metallionen zur Herstellung von Makrozyklen benutzen (Templat-Effekt). Z.B.: Alkali- und Erdalkalimetallionen beschleunigen die Bildung von Benzo[18]crown-6 in Methanol
Stereochemie der oktaedrischen Substitution • Die normale oktaedrische Substitution erfolgt unter Retentionder Konfiguration • Dies deutet darauf hin, dass die normale fünffach koordinierte Zwischenverbindung eine tetragonal pyramidale Struktur beibehält (quadratische Pyramide) • In manchen Fällen ist der Substitutionsvorgang jedoch von einer stereochemischen Umwandlung begleitet, z.B. • trans-[Co(NH3)4Cl2]+ + H2O ergibt 55% cis- und 45% trans-Produkt • trans-[Co(en)2(OH)Cl]+ + H2O ergibt 75% cis und 25% trans-Produkt • trans-[Co(en)2Br2]+ + H2O ergibt 30% cis und 70% trans-Produkt
Das Auftreten stereochemischer Umlagerungen kann durch die Annahme einer trigonal-bipyramidalen Struktur der fünffach koordinierten Zwischenverbindungen erklärt werden. • Der Austritt von X wird durch die Bewegung eines Paares ursprünglich trans zueinander stehender Liganden begleitet, von denen jeder cis zur austretenden Gruppe X steht • Es gibt 2 Möglichkeiten zur Bildung einer trigonalenBipyramide • Die eintretende Gruppe Y tritt entlang einer der trigonalen Kanten ein • 6 mögliche Reaktionswege, 2 davon führen zur Wiederherstellung der relativen Positionen der Liganden im Komplex
Welche Liganden ermöglichen stereochemische Umwandlung? • Voraussetzung ist das Vorhandensein eines einsamen Elektronenpaares in einem Orbital, dessen Symmetrie eine π-Bindung vom Liganden zum Metall ermöglicht, sobald das Umklappen von der quadratischen Pyramide zur trigonalen Bipyramide erfolgt ist: • Stabilisierung des trigonalen ÜZ
Ligandensubstitution in quadratisch-planaren Komplexen • Modellfall: d8 Pt2+ Komplexe („inert“: reagieren langsam) • [Pt(dien)X]+ • X- Halogenidion • dien Diethylentriamin Kinetische Studien an quadratisch-planaren Pt2+ Komplexen ergeben gewöhnlich ein aus zwei Termen bestehendes Geschwindigkeitsgesetz: 2 simultane Mechanismen
Wird in einem Diagramm k gegen [E] aufgetragen, so ergibt sich eine Gerade: • k = k1 + k2 [E] • Der Ordinatenabschnitt k1 ist derselbe für verschiedene E (z.B. Cl- oder Br-) • Der Anstieg k2 hängt sehr stark von der Natur von E ab! • Außerdem ist k2 10-100 mal so groß wie k1. • Substitution in quadratisch-planaren Komplexen vorwiegend durch einen assoziativen Mechanismus.
82Br herstellbar durch Neutroneneinfang aus 81Br t1/2=35.3 h Quelle: Porterfield
aus: William W. Porterfield, Inorganic Chemistry, San Diego, 1993
3 Beweise für den assoziativen Mechanismus • 1. schwacher Effekt der austretenden Gruppe • 2. starker Effekt der eintretenden Gruppe (= geschwindigkeitsbestimmender Schritt) • 3. negative Aktivierungsentropie
Assoziativer Mechanismus für quadratisch-planare Komplexe wahrscheinlicher als für oktaedrische • Koordinationszahl niedriger, Komplex (zumindest potentiell) koordinativ ungesättigt • Die planare Anordnung lässt für einen eintretenden Liganden einen breit gewinkelten Zugang offen (ausser die planaren Liganden sind selbst sehr umfangreich)