Download
1 / 60
600 likes | 1.12k Views
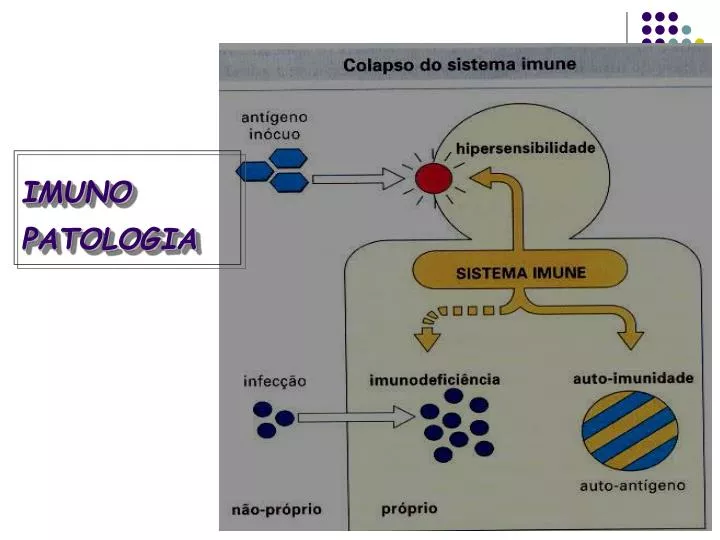
IMUNO PATOLOGIA. IMUNOSSENESCÊNCIA. Declínio da Função imune que ocorre em todos os idosos, fisiologicamente estabelecida, e que não decorre de doença de base, desnutrição, exposição a agente tóxico ou desordem genética. Fatores Genéticos; Doenças concomitantes Estilo de Vida.

E N D
IMUNOSSENESCÊNCIA Declínio da Função imune que ocorre em todos os idosos, fisiologicamente estabelecida, e que não decorre de doença de base, desnutrição, exposição a agente tóxico ou desordem genética. Fatores • Genéticos; • Doenças concomitantes • Estilo de Vida
HIPERSENSIBILIDADE Ag deSUPERFÍCIE TIPO l TIPO ll IgG ALERGENO IgE ALVO MASTÓCITO COMPLEMENTO IMUNE COMPLEXO Ag TIPO lV TIPO lll LINFÓCITO T LINFOCINAS COMPLEMENTO MEMBRANA BASAL MACRÓFAGO MEDIADORES INFLAMATÓRIOS TECIDO
DOENÇAS AUTO-IMUNES • Auto-imunidade: • habilidade de discriminar self (normal do repertório imune) • Controle: auto-tolerância • ausência de resposta imune ou resposta controlada (supressão) para determinado(s) antígeno(s). • Doença Auto-imune: • estado clínico-patológico com resposta (exacerbada) de auto-agressão e lesões teciduais (Antígeno desencadeante é self) • Ag-específica, • adquirida, • mantida (memória). • reconhecimento no contexto moléculas MHC (predisposição genética) • Co-estimulação via interação de receptores celulares e citocinas inflamatórias(associação com processos inflamatórios crônicos)
CARACTERÍSTICAS IMUNOLÓGICAS NA AUTO-IMUNIDADE • Hipergamaglobulinemia • Presença de auto-Ac séricos específicos (testes imunológicos) – Hipersensibilidade tipo II • Infiltrado de linfócitos, plasmócitos e macrófagos no tecido afetado – Hipersensibilidade tipo IV • 4) Depósito dos Imunocomplexos “autoAc-Ag self” em vasos (vasculites), articulações (artrite) e glomérulo renal (nefrite) desencadeando mecanismos de hipersensibilidade tipo III. Auto-Ac são encontrados em baixa concentração em indivíduos sadios.
Ag deSUPERFÍCIE TIPO l TIPO ll IgG ALERGENO IgE ALVO MASTÓCITO COMPLEMENTO IMUNE COMPLEXO Ag TIPO lV TIPO lll LINFÓCITO T LINFOCINAS COMPLEMENTO MEMBRANA BASAL MACRÓFAGO MEDIADORES INFLAMATÓRIOS TECIDO HIPERSENSIBILIDADE
Tireoidite de Hashimoto Mixedema primário Tireotoxicose Anemia perniciosa Gastrite atrófica auto-imune Doença de Adddison Menopausa precoce Diabete mellitus tipo I Síndrome de Goodpasture Miastenia gravis Infertilidade masculina Pênfigo vulgaris Penfigoide Oftalmia sympathetic Uveite facogenica Esclerose multipla (?) Anemia hemolítica auto-imune Púrpura trombocitopênica idiopática Leucopenia idiopática Cirrose biliar primária Hepatite crônica ativa Cirrose criptogênica Colite ulcerativa Síndrome de Sjögren Artrite reumatóide Dermatomiosite Escleroderma Doença mista do tecido conjuntivo Lúpus eritematoso sistêmicodiscoide Lúpus eritematoso sistêmico Órgão específica ESPECTRO DAS DOENÇAS AUTO-IMUNES Não órgão específica Ref. BROSTOFF, J et al., 1991
LÚPUS ERITEMATOSO SISTÊMICO “Doença crônica inflamatória não infecciosa, envolvendo vários órgãos, com episódios de agudização e remissão, apresentando início insidioso ou agudo, e nos tecidos expostos à luz solar gera um exantema eritematoso” Freqüente em mulheres (8:1), jovens (3:1). FATORES PREDISPONENTES Hormônios (estrógeno): puberdade, gestação. Aumento da freqüência de HLA- B8, -B15, DRw2, DRw3. LÚPUS INDUZIDO POR DROGAS Síndrome reversível (LES-like): antiarrítmicos (procainamida), anti-hipertensivos (hidralazina), anti-convulsivantes e clorpromazina. Anticoncepcionais e isoniazida induzem produção de anticorpos anti-núcleo, sem gerar sintomas.
LÚPUS ERITEMATOSO SISTÊMICO Paciente com LES mostrando inflamação e vasculite
LÚPUS ERITEMATOSO SISTÊMICO SINAIS E SINTOMAS rash cutâneo fotossensível, poliartrite (osteonecrose e vasculites) e dermatite (urticária, edema, ulcerações, perda de cabelo).Semelhantes a outras auto-imunidades sistêmicas (esclerose progressiva sistêmica, polimiosite, artrite reumatóide). Manifestações renais e SNC: IC (hipersensibilidade tipo III)
ARTRITE REUMATÓIDE “Doença inflamatória sistêmica crônica progressiva deformante envolvendo tecidos e articulações, a mais freqüente das auto-imunidades” CARACTERÍSTICAS EPIDEMIOLÓGICAS • Incidência: 1% a 2%. Idade: 30 - 50 anos (mulheres) • Associação com HLA-DR4 e –DR1. SINAIS E SINTOMAS Poliartrite destrutiva com vasculite sistêmica. Freqüente início após sinovite com formação de auto-Ac anti-IgG (Fator reumatóide), seguida de processos destrutivos teciduais. Nas juntas articulares a inflamação reduz a lubrificação pelo liquído sinovial (despolimerização do ácido hialurônico). Manifestações extra-articulares: perda de peso, nódulos subcutâneos, vasculites, neuropatias, miopatias, osteoporose.
ARTRITE REUMATÓIDE PATOGENIA Papel importante da resposta T • (ciclosporina A melhora o quadro) • Auto-Ag: colágeno tipo II no tecido sinovial (?). TNF-: papel central na inflamação e Destruição tissular. Anti- TNF-:melhora em alguns casos (infliximabe)
Doenças aparentemente de causa auto-imune 1) Doença de Crohn • inflamação granulomatosa intestinal (íleo e reto). • Extra-intestinal: pele, olhos, articulações, fígado. • Diagnóstico diferencial: diverticulites, apendicite, neoplasia 2) Colite ulcerativa • Mucosa do cólon com inflamação crônica e ulcerações • Ac: anti-células cólon (em alguns ANCA) 3) Doença Celíaca: • Hipersensibilidade a proteínas de grãos/cereais (gliadina) • Ac e LT anti-gliadina (IgA) e anti-endomísio (50% anti-reticulina) • Biópsia do intestino delgado proximal: lâmina própria intestinal com infiltrado de Ly e Pz, associado com atrofia das vilosidades. • Associação com dermatite herpetiforme (coleção subepidérmica de IgA e C, erupções vesiculares de pele e intenso prurido) • Distinguir de alergia alimentar a glúten (IgE-mediada, aguda)
IMUNODEFICIÊNCIAS • Sistema de defesa insuficiente para resposta imune compatível com a vida “normal”. • Pode ser detectada a disfunção. • Classificação: • Quanto à causa: • Primárias ou congênitas [controle genético (?)] • Secundárias ou adquiridas • Quanto ao comprometimento do sistema de defesa • Combinada T e B • Celular • Humoral • Fagocitose/complemento
IMUNODEFICIÊNCIAS SECUNDÁRIAS • drogas: anti-inflamatórias (corticóides), imunossupressoras (ciclosporina, ciclosfosfamida, metotrexate), antibióticos (mitomicina), radiação, hidantoínas • doenças: neoplasias, diabetes, enteropatias, síndromes nefróticas • HIV (AIDS) • estados desnutricionais (proteínas) • infecções: tuberculose, sarampo, varicela, etc (transitórias),
IMUNODEFICIÊNCIAS e INFECÇÕES • DEFICIÊNCIA DE ANTICORPOS, FAGÓCITOS E COMPLEMENTO • * bactérias extracelulares • HUMORAL - S.pneumoniae, S.aureus, H.influenzae, • N.meningitidis, G.lamblia • FAGÓCITOS- S.aureus, S.epidermidis, Serratia sp., E.coli, • Klebsiella sp., Pseudomonas sp. • COMPLEMENTO - Neisseria sp., S.pneumoniae, H.influenzae • DEFICIÊNCIA CELULAR • vírus, fungos, bactérias intracelulares, protozoários, • agentes oportunistas • tumores
AVALIAÇÃO LABORATORIAL DA IMUNOCOMPETÊNCIA in vivo, in vitro e ex vivo • Triagem • Hemograma completo • Eletroforese de proteínas • Dosagem sérica de IgA, IgM e IgG ? Teste de hipersensibilidade tardia com PPD
Imunodeficiência Humoral • Deficiência de IgA • IgA sérica < 7 mg/dL. Outras Ig normais ou aumentadas • Imunidade celular normal ou aumentada • Prevalência 1:1.000 • Associação com doenças alérgicas, gastrointestinais, auto-imunes. • Não fazer uso de gamaglobulina
IMUNIDADE CELULAR • Contagem de LT (80-90% Lcirculantes). • Subpopulações T: CD4+ / CD8+ (2 / 1) • NK CD16/CD56 • Testes de Hipersensibilidade Tardia: função T “in vivo” (candidina, tricofitina) • Biópsia (linfonodos, timo): presença, localização (área paracortical, medula do timo) e semi-quantificação de LT
TRATAMENTO • HIGIENE AMBIENTAL E PESSOAL • Evitar contato com agentes infecciosos • NUTRIÇÃO Aleitamento materno, dieta balanceada • VACINAÇÃO Vírus mortos ou inativados • CITOCINAS • Interferon g - DGC • G-CSF, GM-CSF - neutropenias • TRANSPLANTE DE CÉLULAS PRIMORDIAIS • Medula óssea, cordão umbilical, sangue periférico • TERAPIA GÊNICA • Deficiência de ADA
HIV - AIDS Retrovírus esférico (80-130nm) Duas fitas RNA, Transcriptase reversa Envelope: bicamada lipídica (origem na membrana célula hospedeira) com duas gp (gp160/120/41) HIV-2 gp36 Matriz protéica (core): p17 e capsídeo p24
Infecção pelo HIV Adesão e fusão (gp120 e CD4) + co-receptores (quimiocinas) [CXCR4 LT] ou [CCR5 L, Mo e M] • gp41 interage com co-receptor e ocorre a fusão. • perde envelope, libera RNA, que é transcrito para DNA (TR e ribonuclease H) • DNA viral se integra ao genoma da célula (integrase) = Próvirus • Ciclo de replicação viral: célula é ativada (p.e., por TNF, IL-1, IL-2, IL-6, espécies reativas de oxigênio que ativa fator de transcrição nuclear NF-B) • NF-B no citoplasma, 2 sub-unidades p50 e p65 ligadas à I-B (inibidor) • Subunidades liberadas, p65 e p50 translocam para núcleo e interagem com sítios LTRs do HIV, estimulando transcrição de RNA viral • ribossomos catalisam síntese das proteínas virais, e clivagem é catalisada por protease viral. Primeiras proteínas: tat: transativador de expressão gênica e rev: transporta RNAm do núcleo para o citoplasma.
Imunopatogenia - infecção pelo HIV QUEDA DO NÚMERO DE LINFÓCITOS T CD4+ Mecanismos controversos. • Efeito citopático: • replicação viral e rompimento da membrana celular; • acúmulo de DNA e RNA levando à apoptose; • sincícios (gp120 em cél. infectada e CD4 de cél. vizinha); • Efeito depleção direta: apoptose, resposta alogênica contra LT, citotoxicidade do sistema de defesa (NK, LT CD8+, Ac) apoptose de LT CD4+: o no. de células que morre é grande (2x109/dia), parte não estava infectada.
Infecção pelo HIV - Classificação (CDC, 1994) CATEGORIAS de acordo com condições clínicas (A, B, C) e nº CD4+ [500/L (1); 200 a 500 /L (2); < 200/L (3)] • A: assintomática ou linfadenopatia persistente • B: moderada deficiência com um dos critérios: • condições indicativas de imunodeficiência celular; • evolução complicada pela infecção pelo HIV. (candidíase persistente; diarréia, neuropatia periférica) • C (AIDS): grave e severa deficiência T: criptosporidíase, isosporíase, sarcoma de Kaposi, criptococose, histoplasmose, toxoplasmose, citomegalovirose, candidíase bronquial, linfoma cerebral, etc.
HIV 106 RNA viral 200 AIDS 102 JANELA
Ag deSUPERFÍCIE TIPO l TIPO ll IgG ALERGENO IgE ALVO MASTÓCITO COMPLEMENTO IMUNE COMPLEXO Ag TIPO lV TIPO lll LINFÓCITO T LINFOCINAS COMPLEMENTO MEMBRANA BASAL MACRÓFAGO MEDIADORES INFLAMATÓRIOS TECIDO ALERGIAS E HIPERSENSIBILIDADE
Alergia é uma reação exagerada do organismo contra substâncias inofensivas (respiratórias, alimentares, drogas).
Prevalence, % 35 30 25 20 15 10 5 Year 1960 1970 1980 2000 1990 Aumento da Prevalência da doença alérgica Reference: Strannegård & Strannegård. Allergy 2001;56:91-102
Asma Rinite alimentar Picada insetos Alérgenoscomuns ácaros pólens ácaros, poeira epitélios pólens frutos do mar, ovo, leite, soja, amendoim, peixe Abelhas, vespas, formigas oral Via de entrada: inalatória inalatória subcutânea Resposta inflamação obstrução fadiga falta de ar congestão nasal prurido espirros anafilaxia vômito, dores abdominais, urticária Anafilaxia, edema, vasodilatação local Principais alergias mediadas por IgE
Hipersensibilidade IV – Tardia (Celular) • Ag fagocitado não é eliminado facilmente: Persistência do Ag. • Metais e outras substâncias ligadas em proteínas • Macrófagos ativados liberam citocinas (IFN, IL-2). Ativando mais LTh1 (mais citocinas) • O processo cronifica com fibrose, necrose tecidual, calcificação, granulomas • Leva até 2-4 semanas para formar o granuloma (típico) ou reabsorver o processo • Ex: Dermatite alérgica de contato
Alergia – Tardia (Celular) Dermatite de Contato • na pele os Ags são fagocitados pelas células dendríticas e apresentados a LT. • Reação com infiltrado mononuclear em 12-24hs acompanhado por edema e formação de microvesículas. • Ex: uso de níquel e cobre em bijuterias, neomicina em pomadas, p-fenileno-diamina em laquês, cromatos e picratos. • Perfil tipo Th1
Fisiopatologia da Rejeição de Enxertos Moléculas do MHC (major histocompatibility complex)/ HLA: Human Leucocyte AntigenEstrutura e função /Importância nos transplantes • Transplante – processo de retirada de células, tecidos ou órgãos de um indivíduo e inserção em indivíduo diferente. • Ortotrópico – Inserção em local anatômico habitual • Heterotrópico – Inserção em local anatômico diferente • Enxerto – células, tecidos ou órgãos transplantados • Autólogo – do indivíduo para si mesmo • Isogênico ou singênico – entre indivíduos geneticamente idênticos • Xenogênico – entre indivíduos de espécies diferentes • Alogênico – entre indivíduos não-idênticos da mesma espécie
Imunologia do Transplante - Histórico • Enxertos de pele alogênico (em duas semanas – necrose e desprendimento) • Medawar e Snell – estudo em animais singênicos – identificam moléculas herdadas que são específicas. • Ciclosporina A – bloqueia a ativação do fator de transcrição (fator nuclear de Células T ativadas) que é necessário para transcrição de genes de citocinas (particularmente IL-2). • Investigação • induzir tolerância aos aloantígenos • Evitar interação entre moléculas B7 (células dendríticas do doador) e receptores CD28 (células T do hospedeiro) - proteínas ou Ac anti-B7. • clonagem celular
Doador Receptor Tratamento Rejeição A B Nenhum Lenta A B pré-transplante de A Rápida A B Inóculo de LT de B pré-transplantado de A Rápida C B pré-transplante de A Lenta Memória Imunológica Papel de LT na rejeição do enxerto Especificidade antigênica Peter Medawar
Imunologia do Transplante • O reconhecimento de células transplantadas como próprias ou estranhas é determinado por genes polimórficos herdados de ambos os pais e expressos em co-dominância. • Genes – Complexo principal de histocompatibilidade (CPH/MHC – major histocompatibility complex). • Moléculas – Moléculas do MHC (ou HLA – Human leukocyte antigen?) • Classe I • Classe II • Moléculas de histocompatibilidade secundárias – parecem ser menos imunogênicas.
Rejeição Imunológica a Enxertos • Se os LT são selecionados para serem restritos ao MHC próprio, como as moléculas do MHC (da célula do doador) são apresentadas às células imunes do receptor? • A alorrestrição é por diferenças de polimorfismo – há similaridades
Rejeição Imunológica a Enxertos Abbas & Lichtman. Imunologia Celular e Molecular. 5a ed. Rio de Janeiro:Elsevier. 2005
Rejeição Imunológica a EnxertosAlorreconhe-cimento Abbas & Lichtman. Imunologia Celular e Molecular. 5a ed. Rio de Janeiro:Elsevier. 2005
Rejeição Imunológica a EnxertosAlorreconhecimento • Cada célula alogênica expressa cerca de 105 cópias de cada molécula do MHC. • Reconhecimento da molécula do MHC na célula apresentadora de Ag do doador, ou seja, reação cruzada entre o TCR normal que foi selecionado para reconhecer peptídeos estranhos no contexto MHC próprio, e a molécula MHC alogênica e um peptídeo. • Diferentes peptídeos serão apresentados pela molécula MHC do doador, então múltiplas possibilidades de alorreconhecimento são possíveis.
Mecanismos de destruição do aloenxerto • Rejeição celular • LT CD8+ (Tc): morte celular • LT CD4+ (Th): hipersensibilidade tardia e ativação de LB (Ac) • Reações mediadas por Ac • Hiperaguda: quando o receptor possui Ac anti-HLA do doador • Ac reagem com endotélio vascular do órgão/tecido enxertado (citotoxicidade mediada por Ac) = Vasculite de rejeição
Mecanismos de destruição do aloenxerto Abbas & Lichtman. Imunologia Celular e Molecular. 5a ed. Rio de Janeiro:Elsevier. 2005
Rejeição Aguda(prevenida/controlada por imunossupressores) Dias/semanas pós-transplante em indivíduos não suprimidos Lesão vascular e parenquimatosa • Resposta T: CD4+ e CD8+ (ativados) • CD8+: destruição de células-alvo e células endoteliais vasculares. • CD4+: orquestra respostas HTT e humoral • Citocinas provocam hiperplasia de músculo liso de vasos (espessamento aterosclerótico) + espessamento da íntima por fibroblastos, miócitos e macrófagos espumosos. • Resposta B aguda (vasculite de rejeição) • Lesões necrosantes nos vasos e tecidos do entorno. • Depósitos de IC (inclusive à distância) – citotoxicidade mediada por imunocomplexos (hipersensibilidade tipo III)
Rejeição Aguda Abbas & Lichtman. Imunologia Celular e Molecular. 5a ed. Rio de Janeiro:Elsevier. 2005
Rejeição Crônica (difícil previsão)(adiada por imunossupressores) Fibrose e anormalidades vasculares • Alterações vasculares com fibrose extensa e densa (intersticial e parenquimatosa). • Lúmen dos vasos vai sendo ocupado por células de músculo liso e tecido conjuntivo da íntima do vaso. • O tecido vai fibrosando e perdendo função.
Rejeição Crônica Abbas & Lichtman. Imunologia Celular e Molecular. 5a ed. Rio de Janeiro:Elsevier. 2005