Download

1 / 38
410 likes | 598 Views
ANALISI BIOINFORMATICA DELLE PROTEINE. Matteo Ramazzotti matteo.ramazzotti@unifi.it. Programma.

E N D
ANALISI BIOINFORMATICA DELLE PROTEINE Matteo Ramazzotti matteo.ramazzotti@unifi.it
Programma Banche dati proteiche. Interrogazione delle banche dati. Allineamento di sequenze proteiche. Matrici di sostituzione. Allineamento con gap. Allineamento globale e locale. BLAST e FASTA. Allineamento multiplo. Visualizzazione degli allineamenti. Applicazioni dei profili di multiallinemento. Ricerca di pattern e motivi funzionali nelle proteine. Banche dati dei profili proteici. Filogenesi molecolare. L’orologio molecolare. Analisi della struttura primaria delle proteine. Struttura secondaria delle proteine. Strumenti per la predizione della struttura secondaria. Visualizzazione tridimensionale delle proteine. Strumenti per la proteomica. RasMol. Swiss PDB Viewer. Strumenti per la predizione della struttura tridimensionale. Homology modelling. Interazione tra proteine. Docking. Testi consigliati: G.Valle et al. – “Introduzione alla bioinformatica” – Zanichelli A.Tramontano – “Bioinformatica” - Zanichelli
Cos’è la bioinformatica E’ una scienza multidisciplinare che integra conoscenze informatiche, chimiche, matematiche, biologiche allo scopo di collezionare ed elaborare sistematicamente ogni informazione per ottenere la massima resa dalle ricerche sperimentali ma anche per sviluppare queste ultime in modo più mirato. Grazie alla bioinformatica è possibile avere uno sguardo d’insieme su tutte le conoscenze scientifiche e da questo partire per ulteriori osservazioni sia mediante metodi informatici, sia mediante metodi sperimentali.
Chi si occupa di bioinformatica può appartenere a due categorie principali:SVILUPPATORI: coloro che si occupano di creare nuovi strumenti informatici per l’analisi scientifica UTENTI: coloro che utilizzano gli strumenti bioinformatici per ottenere dati e da questi partire per l’analisi sperimentale vera e propria. • il programma che si utilizza può definirsi il metodo sperimantale della bioinformatica • le banche dati posso definirsi il materiale sperimentale utilizzato dalla bioinformatica
Banche dati Si possono raggruppare in quattro categorie principali: • di biosequenze (dette anche primarie) • genomiche • di espressione genica • proteiche Nel corso verranno trattate le banche dati proteiche anche se non bisogna dimenticare che molti dati sulle proteine derivano dalle banche dati primarie, cioè quelle nucleotidiche.
Traduzione in silico Data una sequenza nucleotidica è possibile tradurla utilizzando tutti e tre i frame di lettura (+1, +2, +3) di entrambi i filamenti (senso e antisenso), allo scopo di individuare una Open Reading Frame (ORF)cioè una sequenza proteica di lungheza adatta ad essere una proteina (non meno di 70-100 residui). In questo modo, e con programmi appositi che scansionano tutto un genoma, è possibile trovare tantissime proteine PUTATIVE, cioè possibili ma non verificate. +2 T R M P L S L S M G M P I V H I H S N A N A S L S F D G Y A N C P H S L E R E C L S L F R W V C Q L S T F T R 1 gaacgcgaatgcctctctctctttcgatgggtatgccaattgtccacattcactcgt F A F A E R E K S P Y A L Q G C E S T R S H R E R K R H T H W N D V N V R V R I G R E R E I P I G I T W M * E +1 +3
Alanine Ala A GC[CATG] Cysteine Cys C TG[CT] Aspartic AciD Asp D GA[CT] Glutamic Acid Glu E GA[AG] Phenylalanine Phe F TT[CT] Glycine Gly G GG[CATG] Histidine His H CA[CT] Isoleucine Ile I AT[CAT] Lysine Lys K AA[AG] Leucine Leu L CT[CATG], TT[AG] Methionine Met M ATG AsparagiNe Asn N AA[CT] Proline Pro P CC[CATG] Glutamine Gln Q CA[AG] ARginine Arg R CG[CATG], AG[AG] Serine Ser S TC[CTAG], AG[CT] Threonine Thr T AC[CATG] Valine Val V GT[CATG] Tryptophan Trp W TGG TYrosine Tyr Y TA[CT] STOP - - TA[AG], TGA Il codice genetico Si definisce DEGENERATO eRIDONDANTE. E’ composto da 64 diversi codoni che codificano i 20 amino acidi. La tebella accanto mostra le varie degenerazioni dei codoni per ogni amino acido.
Aminoacil-tRNA tRNA rRNA mRNA ------------RBS----ATG AAA TAC Trascrizione TAA Sintesi proteica Struttura primaria Struttura secondaria Folding Struttura terziaria
Gli amino acidi Sono composti organici che presentano almeno un gruppo carbossilico (-COOH) a funzione acida e un gruppo aminico (-NH2) a funzione basica Le proteine sono composte soltanto da alfa-amino acidi, legati tra loro mediante legami detti PEPTIDICI che si instaurano tra il COOH e l’NH2. Ciò che diversifica i vari amino acidi è la catena laterale legata al carbonio alfa, che può conferire all’amino acido caratteristiche chimico-fisiche diverse.
Idrofobici Basici Aromatici Polari non carichi Acidi Strutturali In base alla catena laterale si riconoscono 5 gruppi principali di amino acidi
I polimeri di alfa amino acidi (le proteine) sono influenzati dalle caratteristiche chimico-fisiche delle catane laterali e in base a principi di interazioni deboli di tipo idrofobico o elettrostatico si osservano dei ripiegamenti, fino al raggiungimento della minor energia termodinamica. Questo processo, denominato FOLDING, è alla base del funzionamento delle proteine, visto che solo se sono correttamente strutturate esse assumeranno la loro forma e soprattutto FUNZIONE definitiva. La strutturazione delle proteine dipende quindi principalmente dalla sequenza dei residui che la compongono, oltre che dall’ambiente in cui si strutturano
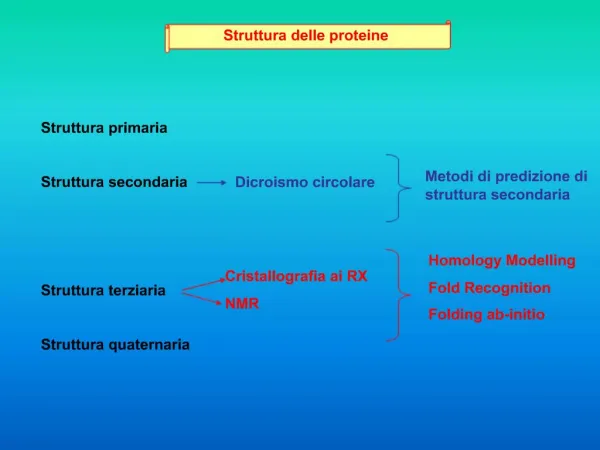
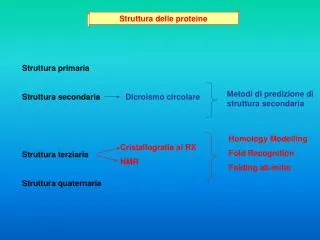
Struttura delle proteine Il legame peptidico ha delle caratteristiche di doppio legame e costringe i due atomo adiacenti a giacere sullo stesso piano. La rotazione della molecola avviene intorno al carbonio alfa, ma non tutti gli angoli di rotazione sono permessi a causa degli ingombri sterici delle diverse catene laterali e dello scheletro stesso.
Il legame peptidico genera una polarità negli scheletri proteici per cui si vengono a formare PONTI IDROGENO tra gli idrogeni dei gruppi amidici e ossigeni dei carbonili. Queste interazioni deboli portano la struttura primaria della proteina (la sequenza dei suoi residui) a ripiegarsi in una STRUTTURA SECONDARIA in cui sono riconoscibili due formazioni 1- Alfa elica: struttura compatta avvolta in cui i legami idrogeno sono disposti parallelamente allo scheletro. 2 - Beta-strand: struttura rilassata in cui i ponti idrogeno si stabiliscono tra catene adiacenti che possono essere parallele o antiparallele, a formare dei foglietti beta.
Le varie strutture secondarie si collegano tra loro mediante anse (loop) in cui non ci sono ponti idrogeno intramolecolari e che quindi non hanno un’organizzazione definita. In realtà alcune connessioni tra strutture secondarie sono conservate: es. per congiungere due beta-strands antiparalleli serve un connettore che permetta una curva molto stretta. La connessione HAIRPIN LOOP è un esempio. Strutture secondarie che si uniscono formano molto spesso delle strutture super-secondarie denominate MOTIVI
Beta-turn-beta up-down greca jelly-roll super-barrel Alpha-turn-alpha coiled-coil four helix bundle beta-alpha-beta fold di Rossmann
Alcune strutture terziarie Dominio doppio Dominio singolo Dominio quadruplo Dominio triplo
Alcune osservazioni importanti • nello scrivere le proteine o i nucleotidi, usate sempre il carattere COURIER, non altri caratteri con spaziatura ineguale come il times, altrimenti succede questo: VILMAanzichè:VILMA e si perdono gli allineamenti VLLMA VLLMA • utilizzate sempre il blocco note o simili per editare le sequenze, in modo da non avere formattazioni impreviste. Il formato SOLO TESTO è il più adatto. • non copiate MAI a mano le sequenze, anche se brevi. Usate sempre il copia e incolla. Quindi: tenete i dati in formato elettronico, non cartaceo. • per modificare l’aspetto delle sequenze, utilizzate programmi appositi, non fate nulla a mano. Un ottimo programma è il Sequence Manipulation Suite (SMS)
Nascita delle banche dati Inizio anni 70: nasce la tecnologia del DNA ricombinante, che permette di manipolare le sequenze nucleotidiche e di capire la struttura, la funzione e l’organizzazione del DNA. Fine anni 70: pubblicazione dei primi dati genomici, con le prime sequenze nucleotidiche codificanti liberamente accessibili attraverso i rudimenti della rete disponibili a quel tempo tra le varie università. 2001: il Consorzio Pubblico Internazionale e la Celera Genomics forniscono dati del genoma umano completo, aprendo la strada ai progetti di sequenziamento a tappeto. Successivamente, l’approccio biotecnologico ha fornito una serie imponente di dati di natura proteomica grazie all’analisi spettrometrica e all’elettroforesi 2-D, ed una serie altrettanto vasta di dati di trascrittomica grazie alla tecnologia dei microarrays.
Insieme ai dati nasce l’esigenza di sistemi di archiviazione e di ritrovamento facili e esaustivi, in modo da averli a disposizione in ogni istante, dato che sebbene ci siano tantissime informazioni, ognuna deve essere validata e confermata, essendo per la maggior parte dati grezzi non rielaborati. Conoscere il dato non significa capire il dato, serve sempre un approccio sperimentale classico perchè questo sia veramente verificato. => una banca dati è il posto dove cercare i dati da cui partire per una ricerca, non il suo punto di arrivo.
I pionieri 1965: Margareth Dayhoff compila un atlante di proteine omologhe studiando le relazioni tra le sequenze primarie 1970: l’atlante viene reso pubblico in versione elettronica nella banca dati NBRF • nascita della prima banca dati proteica. Ancora non ci sono dati di sequenziamento nucleotidico nella banca, sono tutti dati di natura biochimica classica, mal’idea di rendere disponibili in modo libero dei dati accumulati e organizzati è alla base del concetto che muove gli organizzatori e i curatori delle banche dati, e che muove anche i fondi per la loro gestione
Banche dati primarie 1981: nasce nel Laboratorio Europeo di Biologia Molecolare ad Heidelberg (Germania) l’EMBL-datalibrary, 519 entries con sequenze di DNA e RNA, autore Kurt Stueber 1982: nasce una banca dati simile negli USA, darà vità alla GenBank, autore Walter Goad 1986: nel National Institute of Genetics in Mishima (Giappone) nasce un mirror della GenBank, la DDBJ EMBL GenBank DDBJ => circa le stesse informazioni, organizzate in modo diverso
Infrastrutture principali EMBNet, nata nel 1988 come rete europea a supporto della ricerca bio-molecolare, oggi conta 41 nodi nazionali in paesi europei ed extraeuropei (In Italia il nodo è a Bari) APBioNet (Asian-Pacific Biologic Network), recentemente gemelleta con EMBNet, organizzazione analoga Oggi i due database primari più importanti sono nei centri EBI(Cambridge, UK): EMBL data-library NCBI(USA): GenBank
Organizzazione di un database biologico L’oggetto principale è la ENTRY, una unità riconoscibile grazie ad un identificatore univoco, che possiede una descrizione organizzata in campi standardizzati riconoscibili grazie ad HEADERS univoci nella banca dati. es. Identificatore ----------------- Autore ----------------- Data ----------------- ecc. Ogni banca dati presenta 2 versioni delle entries: Flat-file: un file di testo semplice, formattato, non interattivo HTML (o XML): interattivo, di facile consultazione
L’interattività ha un ruolo centrale per una banca dati, perchè permette di navigare tra le sue entries e quelle di altri databases • sia i flat-file sia le pagine XML sono ricchi di cross-references, riferimenti che mandano ad altre banche dati generiche o specializzate. Si ottiene così per ogni entry una serie di informazioni spesso ridondanti, tra cui è bene sapresi orientare, anche perchè alcune sembrano in contraddizione, es. • una proteina può avere dei riferimenti a sequenze codificanti diverse • una entry può avere più nomi per descriverla o può corrispondere a più autori
Un esempio di entry proteica EMBL (flat-file) 1: AAC74054. orf, hypothetical...[gi:1787203] LOCUS AAC74054 92 aa linear BCT 01-DEC-2000 DEFINITION orf, hypothetical protein [Escherichia coli K12]. ACCESSION AAC74054 VERSION AAC74054.1 GI:1787203 DBSOURCE locus AE000199 accession AE000199.1 KEYWORDS . SOURCE Escherichia coli K12. ORGANISM Escherichia coli K12 Bacteria; Proteobacteria; gamma subdivision; Enterobacteriaceae; Escherichia. REFERENCE 1 (residues 1 to 92) AUTHORS Blattner,F.R., Plunkett,G. III, Bloch,C.A., Perna,N.T., Burland,V., Riley,M., Collado-Vides,J., Glasner,J.D., Rode,C.K., Mayhew,G.F., Gregor,J., Davis,N.W., Kirkpatrick,H.A., Goeden,M.A., Rose,D.J., Mau,B. and Shao,Y. TITLE The complete genome sequence of Escherichia coli K-12 JOURNAL Science 277 (5331), 1453-1474 (1997) MEDLINE 97426617 PUBMED 9278503
REFERENCE 2 (residues 1 to 92) AUTHORS Blattner,F.R. TITLE Direct Submission JOURNAL Submitted (16-JAN-1997) Guy Plunkett III, Laboratory of Genetics, University of Wisconsin, 445 Henry Mall, Madison, WI 53706, USA. Email: ecoli@genetics.wisc.edu Phone: 608-262-2534 Fax: 608-263-7459 REFERENCE 3 (residues 1 to 92) AUTHORS Blattner,F.R. TITLE Direct Submission JOURNAL Submitted (02-SEP-1997) Guy Plunkett III, Laboratory of Genetics, University of Wisconsin, 445 Henry Mall, Madison, WI 53706, USA. Email: ecoli@genetics.wisc.edu Phone: 608-262-2534 Fax: 608-263-7459 REFERENCE 4 (residues 1 to 92) AUTHORS Plunkett,G. III. TITLE Direct Submission JOURNAL Submitted (13-OCT-1998) Laboratory of Genetics, University of Wisconsin, 445 Henry Mall, Madison, WI 53706, USA COMMENT This sequence was determined by the E. coli Genome Project at the University of Wisconsin-Madison (Frederick R. Blattner, director). Supported by NIH grants HG00301 and HG01428 (from the Human Genome
Project and NCHGR). The entire sequence was independently determined from E. coli K12 strain MG1655. Predicted open reading frames were determined using GeneMark software, kindly supplied by Mark Borodovsky, Georgia Institute of Technology, Atlanta, GA, 30332 [e-mail: mark@amber.gatech.edu]. Open reading frames that have been correlated with genetic loci are being annotated with CG Site Nos., unique ID nos. for the genes in the E. coli Genetic Stock Center (CGSC) database at Yale University, kindly supplied by Mary Berlyn. A public version of the database is accessible (http://cgsc.biology.yale.edu). Annotation of the genome is an ongoing task whose goal is to make the genome sequence more useful by correlating it with other data. Comments to the authors are appreciated. Updated information will be available at the E. coli Genome Project's World Wide Web site (http://www.genetics.wisc.edu). *** The E. coli K12 sequence and its annotations are periodically updated; this is version M54. No sequence changes. Annotation updates: updated gene identifications and products; all new functional assignments courtesy of Monica Riley; added promoters, protein binding sites, and repeated sequences described in reference 1. The unique numeric identifiers beginning with a lowercase 'b' assigned to each gene (protein- or RNA-encoding) are now designated as gene synonyms instead of labels. This should allow them to be searched for in Entrez as gene names.
Method: conceptual translation. FEATURES Location/Qualifiers source 1..92 /organism="Escherichia coli K12" /strain="K12" /sub_strain="MG1655" /db_xref="taxon:83333" Protein 1..92 /function="orf; Unknown" CDS 1..92 /gene="b0968" /coded_by="AE000199.1:121..399" /note="o93; 44 pct identical (2 gaps) to 85 residues from acylphosphatase, organ-common type isozyme, ACYO_CHICK SW:P07032 (98 aa)" /transl_table=11 ORIGIN 1 mskvciiawv ygrvqgvgfr yttqyeakrl gltgyaknld dgsvevvacg eegqveklmq 61 wlksggprsa rvervlseph hpsgeltdfr ir // Revised: July 5, 2002.
Ogni banca dati ha dei suoi codici di identificazione e definisce le sue entries secondo un rigido standard, imponendo a priori un certo numero di possibili campi contrassegnati da tag specifici. Nell’esempio visto prima: ACCESSION AAC74054indica il numero di accesso,ORGANISM Escherichia coli K12 Bacteria; Proteobacteria; gamma subdivision; Enterobacteriaceae; Escherichia.indica l’organismo a cui appartiene e la sua tassonomia. Qualsiasi cosa è standardizzata, dai tags agli spazi ed ai segni di punteggiatura. Questo permette ai programmi di RETRIEVAL, cioè di ricerca, di trovare rapidamente ciò che si cerca.
Banche dati proteiche più utilizzate UniProt raccoglie le informazioni dei database Swiss-prot, TrEMBL e PIR. Offre la possibilità di effettuare Text Search o Blast Search. Viene curato anche un database NON RIDONDANTE (UniRef). Molto curato e dattagliato, con annotazioni circa funzione, struttura, modificazioni e altre informazioni utili E’ la traduzione in silico di ogni entry codificante del database primario dell’EMBL, non è accurato, ma è ricchissimo E’ il discendente diretto del database della Dayhoff, è curato a mano e le annotazioni sono molto ricche e precise
Banche dati proteiche più utilizzate E’ un database di famiglie e domini proteici comprensiva di pattern e motivi (signatures) che identificano e rendono riconoscibili e classificabili le proteine. La ricerca in prosite comprende anche altri database strutturali e di classificazione. una signature formattata, definita anche pattern.
Banche dati proteiche più utilizzate Pfam è una raccolta di proteine allineate e di profili generati con gli HMM che descrivono quasi tutte le famiglie e i domini proteici conosciuti. Da qui è possibile una analisi dettagliata sfruttando le risorse disponibili nel server del Sanger Institute per l’analisi familiare delle proteine. Il Proteome Analysis Database è una immensa raccolta di proteine catalogate per organismo di appartenenza e permette analisi interproteomiche mediante opportuni programmi di confronto.
Banche dati proteiche più utilizzate Database di Protein Fingerprints, cioè pattern caratteristici di certe famiglie proteihce Database di domini proteici generato in modo automatico da Swiss-Prot e TrEMBL Database di architetture proteiche annotate per organismo e per famiglia Database di strutture tridimensionali di proteiene altre componenti proteiche